

Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters
- PMID: 27863249
- PMCID: PMC5123897
- DOI: 10.1016/j.cell.2016.09.037
Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters
Abstract
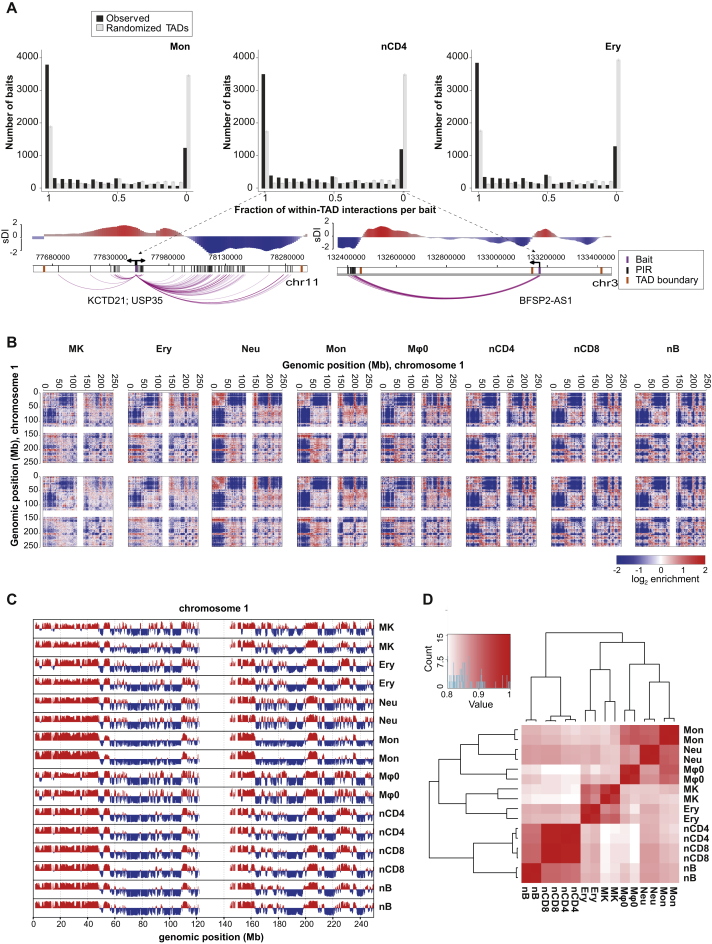
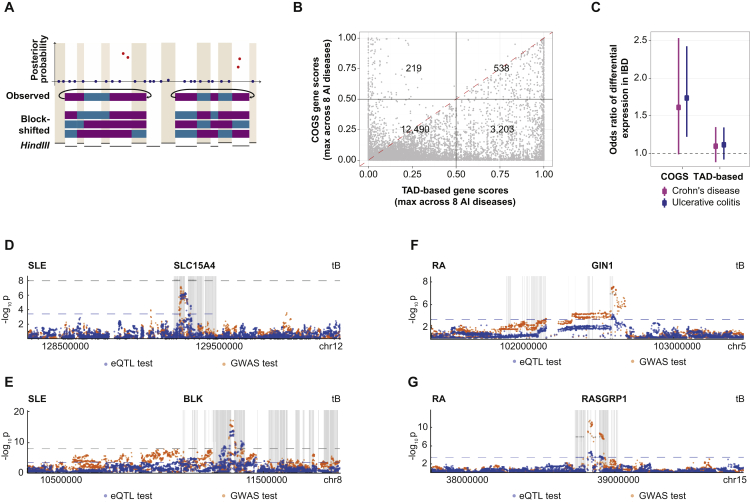
Long-range interactions between regulatory elements and gene promoters play key roles in transcriptional regulation. The vast majority of interactions are uncharted, constituting a major missing link in understanding genome control. Here, we use promoter capture Hi-C to identify interacting regions of 31,253 promoters in 17 human primary hematopoietic cell types. We show that promoter interactions are highly cell type specific and enriched for links between active promoters and epigenetically marked enhancers. Promoter interactomes reflect lineage relationships of the hematopoietic tree, consistent with dynamic remodeling of nuclear architecture during differentiation. Interacting regions are enriched in genetic variants linked with altered expression of genes they contact, highlighting their functional role. We exploit this rich resource to connect non-coding disease variants to putative target promoters, prioritizing thousands of disease-candidate genes and implicating disease pathways. Our results demonstrate the power of primary cell promoter interactomes to reveal insights into genomic regulatory mechanisms underlying common diseases.
Keywords: chromosome conformation; disease gene prioritization; gene regulation; non-coding genetic variation; promoter capture Hi-C.
Copyright © 2016 The Authors. Published by Elsevier Inc. All rights reserved.
Figures











References
-
- Barrett J.C., Clayton D.G., Concannon P., Akolkar B., Cooper J.D., Erlich H.A., Julier C., Morahan G., Nerup J., Nierras C., Type 1 Diabetes Genetics Consortium Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009;41:703–707. - PMC - PubMed
-
- Bentham J., Morris D.L., Cunninghame Graham D.S., Pinder C.L., Tombleson P., Behrens T.W., Martín J., Fairfax B.P., Knight J.C., Chen L. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 2015;47:1457–1464. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
