

TSC loss distorts DNA replication programme and sensitises cells to genotoxic stress
- PMID: 27863419
- PMCID: PMC5356742
- DOI: 10.18632/oncotarget.13378
TSC loss distorts DNA replication programme and sensitises cells to genotoxic stress
Abstract
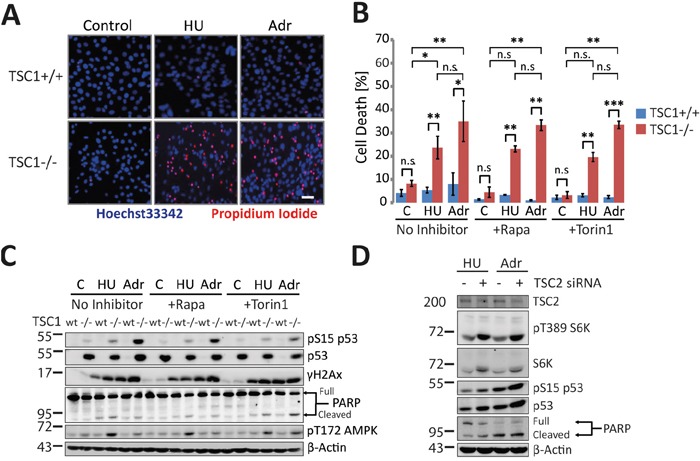
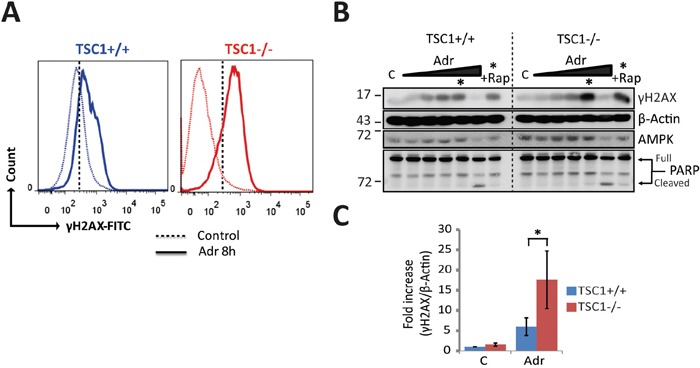
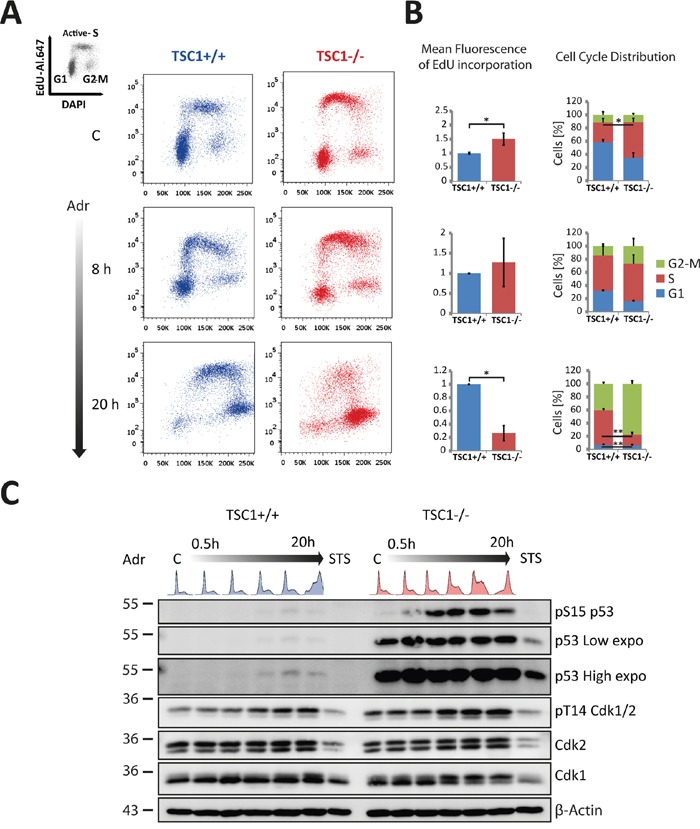
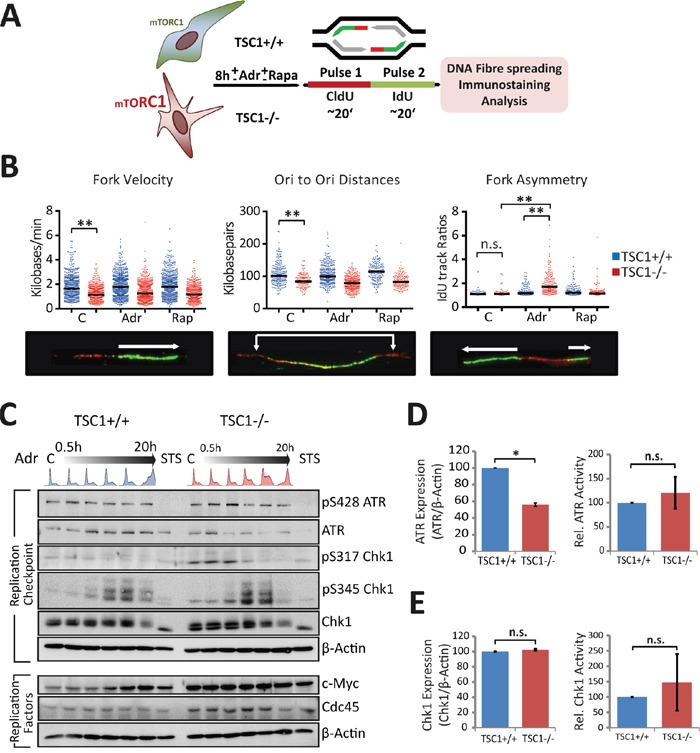
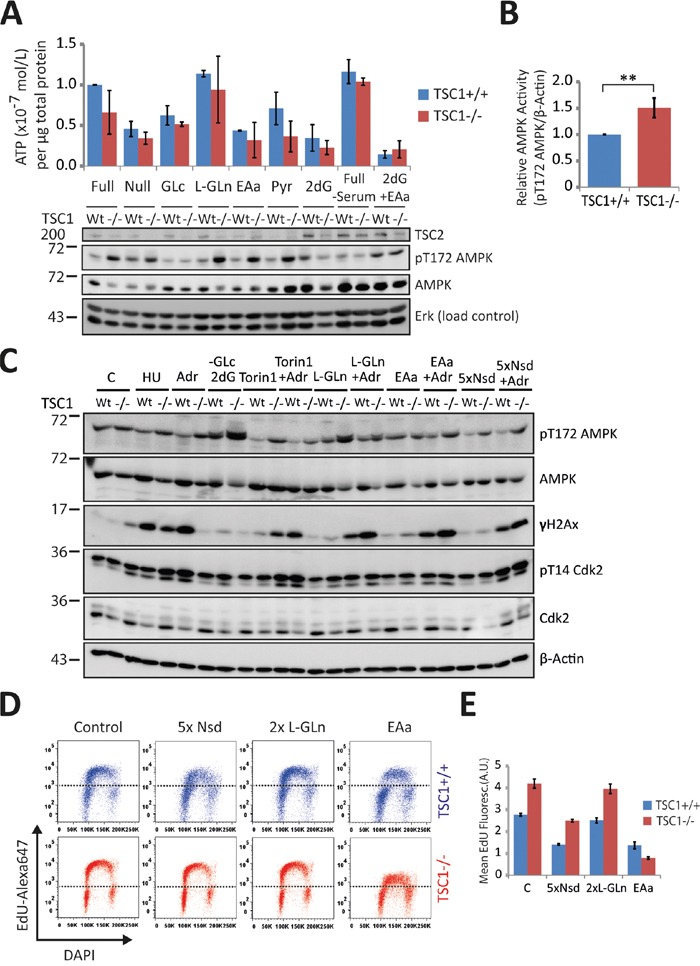
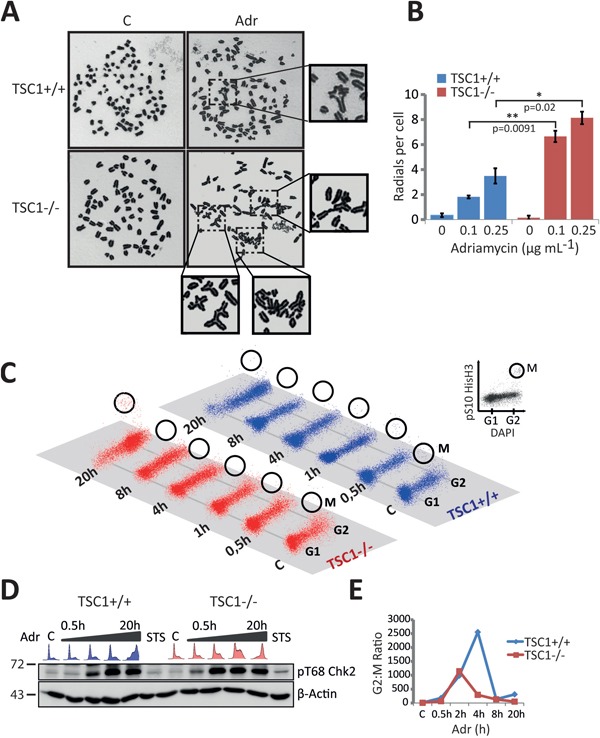
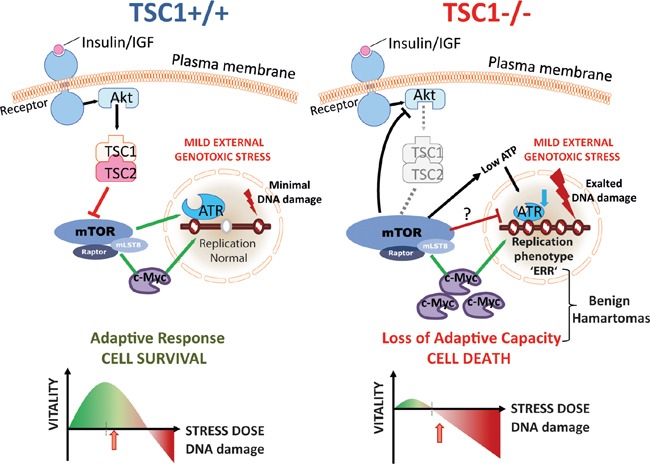
Tuberous Sclerosis (TSC) is characterized by exorbitant mTORC1 signalling and manifests as non-malignant, apoptosis-prone neoplasia. Previous reports have shown that TSC-/- cells are highly susceptible to mild, innocuous doses of genotoxic stress, which drive TSC-/- cells into apoptotic death. It has been argued that this hypersensitivity to stress derives from a metabolic/energetic shortfall in TSC-/- cells, but how metabolic dysregulation affects the DNA damage response and cell cycle alterations in TSC-/- cells exposed to genotoxic stress is not understood. We report here the occurrence of futile checkpoint responses and an unusual type of replicative stress (RS) in TSC1-/- fibroblasts exposed to low-dose genotoxins. This RS is characterized by elevated nucleotide incorporation rates despite only modest origin over-firing. Strikingly, an increased propensity for asymmetric fork progression and profuse chromosomal aberrations upon mild DNA damage confirmed that TSC loss indeed proved detrimental to stress adaptation. We conclude that low stress tolerance of TSC-/- cells manifests at the level of DNA replication control, imposing strong negative selection on genomic instability that could in turn detain TSC-mutant tumours benign.
Keywords: adaptive responses; genotoxic stress; mTORC1; replication stress; tuberous sclerosis.
Conflict of interest statement
All authors declare no competing financial interests or elsewise conflict of interests related to this work.
Figures
Similar articles
-
Rapamycin requires AMPK activity and p27 expression for promoting autophagy-dependent Tsc2-null cell survival.Biochim Biophys Acta. 2016 Jun;1863(6 Pt A):1200-7. doi: 10.1016/j.bbamcr.2016.03.009. Epub 2016 Mar 11. Biochim Biophys Acta. 2016. PMID: 26975583
-
Differentiating the mTOR inhibitors everolimus and sirolimus in the treatment of tuberous sclerosis complex.Neuro Oncol. 2015 Dec;17(12):1550-9. doi: 10.1093/neuonc/nov152. Epub 2015 Aug 19. Neuro Oncol. 2015. PMID: 26289591 Free PMC article. Review.
-
Renal tumours in a Tsc1+/- mouse model show epigenetic suppression of organic cation transporters Slc22a1, Slc22a2 and Slc22a3, and do not respond to metformin.Eur J Cancer. 2013 Apr;49(6):1479-90. doi: 10.1016/j.ejca.2012.10.027. Epub 2012 Dec 7. Eur J Cancer. 2013. PMID: 23228442
-
PAK2 is an effector of TSC1/2 signaling independent of mTOR and a potential therapeutic target for Tuberous Sclerosis Complex.Sci Rep. 2015 Sep 28;5:14534. doi: 10.1038/srep14534. Sci Rep. 2015. PMID: 26412398 Free PMC article.
-
DNA replication stress: from molecular mechanisms to human disease.Chromosoma. 2017 Feb;126(1):1-15. doi: 10.1007/s00412-016-0573-x. Epub 2016 Jan 21. Chromosoma. 2017. PMID: 26797216 Review.
Cited by
-
mTORC1 Is Not Principally Involved in the Induction of Human Endotoxin Tolerance.Front Immunol. 2020 Aug 7;11:1515. doi: 10.3389/fimmu.2020.01515. eCollection 2020. Front Immunol. 2020. PMID: 32849516 Free PMC article.
-
Genome-wide siRNA screens identify RBBP9 function as a potential target in Fanconi anaemia-deficient head-and-neck squamous cell carcinoma.Commun Biol. 2023 Jan 13;6(1):37. doi: 10.1038/s42003-022-04389-3. Commun Biol. 2023. PMID: 36639418 Free PMC article.
-
The GATOR complex regulates an essential response to meiotic double-stranded breaks in Drosophila.Elife. 2019 Oct 25;8:e42149. doi: 10.7554/eLife.42149. Elife. 2019. PMID: 31650955 Free PMC article.
-
Heterozygous loss of TSC2 alters p53 signaling and human stem cell reprogramming.Hum Mol Genet. 2017 Dec 1;26(23):4629-4641. doi: 10.1093/hmg/ddx345. Hum Mol Genet. 2017. PMID: 28973543 Free PMC article.
References
-
- Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell. 2000;100:57–70. - PubMed
-
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. - PubMed
-
- Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. - PubMed
-
- Hengstschläger M, Rodman DM, Miloloza A, Hengstschläger-Ottnad E, Rosner M, Kubista M. Tuberous sclerosis gene products in proliferation control. Mutation research. 2001;488:233–239. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
