

The ChIP-Seq tools and web server: a resource for analyzing ChIP-seq and other types of genomic data
- PMID: 27863463
- PMCID: PMC5116162
- DOI: 10.1186/s12864-016-3288-8
The ChIP-Seq tools and web server: a resource for analyzing ChIP-seq and other types of genomic data
Abstract
Background: ChIP-seq and related high-throughput chromatin profilig assays generate ever increasing volumes of highly valuable biological data. To make sense out of it, biologists need versatile, efficient and user-friendly tools for access, visualization and itegrative analysis of such data.
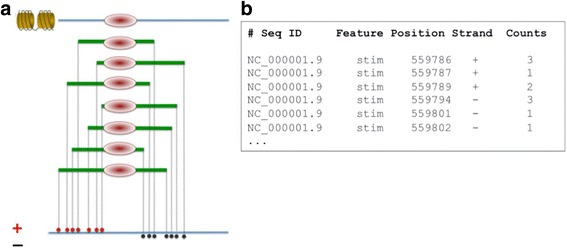
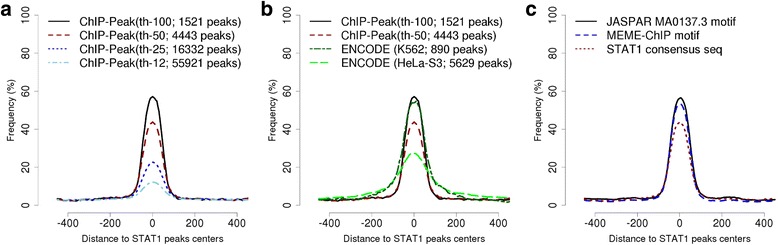
Results: Here we present the ChIP-Seq command line tools and web server, implementing basic algorithms for ChIP-seq data analysis starting with a read alignment file. The tools are optimized for memory-efficiency and speed thus allowing for processing of large data volumes on inexpensive hardware. The web interface provides access to a large database of public data. The ChIP-Seq tools have a modular and interoperable design in that the output from one application can serve as input to another one. Complex and innovative tasks can thus be achieved by running several tools in a cascade.
Conclusions: The various ChIP-Seq command line tools and web services either complement or compare favorably to related bioinformatics resources in terms of computational efficiency, ease of access to public data and interoperability with other web-based tools. The ChIP-Seq server is accessible at http://ccg.vital-it.ch/chipseq/ .
Keywords: Bioinformatics resources; ChIP-seq data analysis; DNA sequence motifs; Genomic context analysis; Histone modifications; Peak finding; Transcription factor binding sites; Web server.
Figures





References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
