

Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy
- PMID: 27866851
- PMCID: PMC5675554
- DOI: 10.1016/j.ccell.2016.10.006
Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy
Abstract
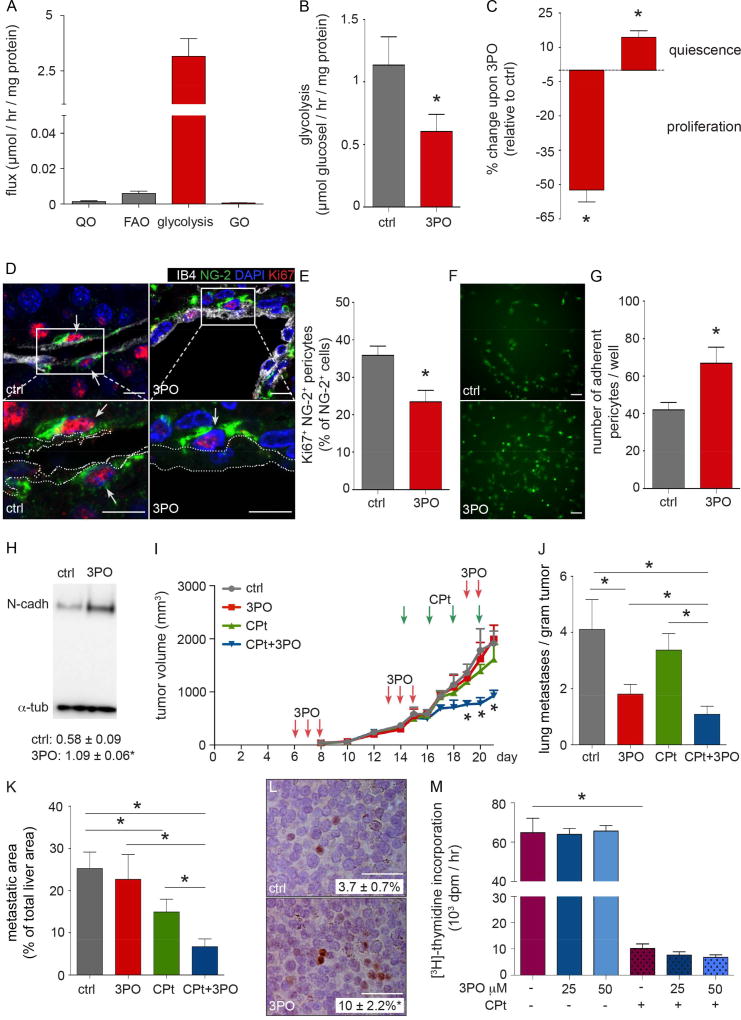
Abnormal tumor vessels promote metastasis and impair chemotherapy. Hence, tumor vessel normalization (TVN) is emerging as an anti-cancer treatment. Here, we show that tumor endothelial cells (ECs) have a hyper-glycolytic metabolism, shunting intermediates to nucleotide synthesis. EC haplo-deficiency or blockade of the glycolytic activator PFKFB3 did not affect tumor growth, but reduced cancer cell invasion, intravasation, and metastasis by normalizing tumor vessels, which improved vessel maturation and perfusion. Mechanistically, PFKFB3 inhibition tightened the vascular barrier by reducing VE-cadherin endocytosis in ECs, and rendering pericytes more quiescent and adhesive (via upregulation of N-cadherin) through glycolysis reduction; it also lowered the expression of cancer cell adhesion molecules in ECs by decreasing NF-κB signaling. PFKFB3-blockade treatment also improved chemotherapy of primary and metastatic tumors.
Keywords: angiogenesis; chemotherapy; glycolysis; metastasis; tumor endothelial cell metabolism; tumor vessel normalization.
Copyright © 2016 Elsevier Inc. All rights reserved.
Conflict of interest statement
The other authors declare no conflict of interests.
Figures







Comment in
-
Targeting PFKFB3 in the Endothelium for Cancer Therapy.Trends Mol Med. 2017 Mar;23(3):197-200. doi: 10.1016/j.molmed.2017.01.008. Epub 2017 Feb 5. Trends Mol Med. 2017. PMID: 28173976
Comment on
-
To PFKFB3 or Not to PFKFB3, That Is the Question.Cancer Cell. 2016 Dec 12;30(6):831. doi: 10.1016/j.ccell.2016.11.007. Cancer Cell. 2016. PMID: 27960081
References
-
- Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Developmental cell. 2011;21:193–215. - PubMed
-
- Benedito R, Roca C, Sorensen I, Adams S, Gossler A, Fruttiger M, Adams RH. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137:1124–1135. - PubMed
-
- Blindt R, Bosserhoff AK, Dammers J, Krott N, Demircan L, Hoffmann R, Hanrath P, Weber C, Vogt F. Downregulation of N-cadherin in the neointima stimulates migration of smooth muscle cells by RhoA deactivation. Cardiovasc Res. 2004;62:212–222. - PubMed
-
- Bogetto L, Gabriele E, Cariati R, Dolcetti R, Spessotto P, Doglioni C, Boiocchi M, Perris R, Colombatti A. Bidirectional induction of the cognate receptor-ligand alpha4/VCAM-1 pair defines a novel mechanism of tumor intravasation. Blood. 2000;95:2397–2406. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
