

Pericyte MyD88 and IRAK4 control inflammatory and fibrotic responses to tissue injury
- PMID: 27869651
- PMCID: PMC5199713
- DOI: 10.1172/JCI87532
Pericyte MyD88 and IRAK4 control inflammatory and fibrotic responses to tissue injury
Abstract
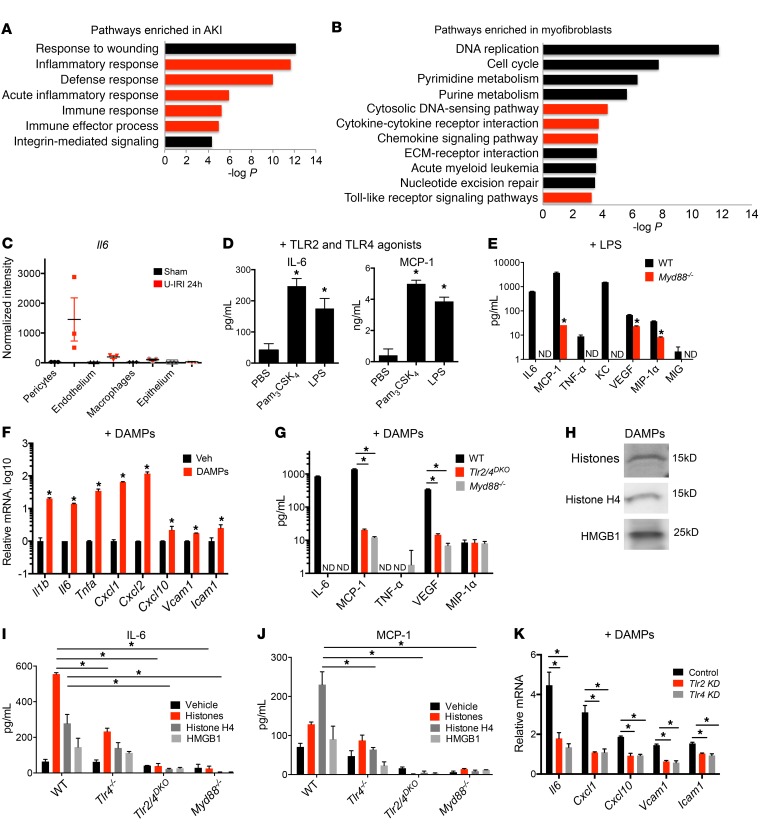
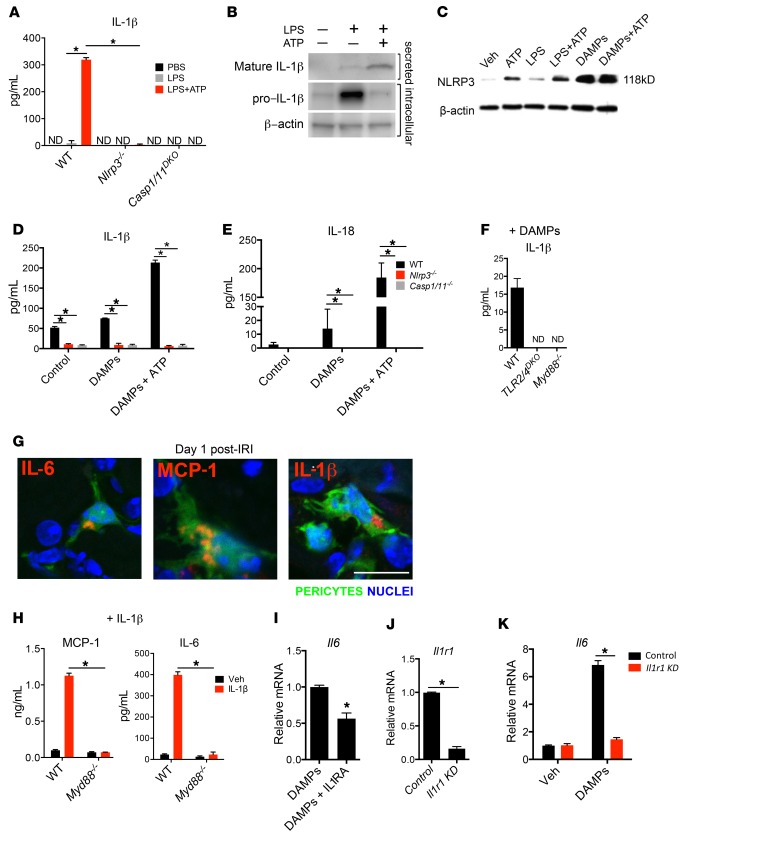
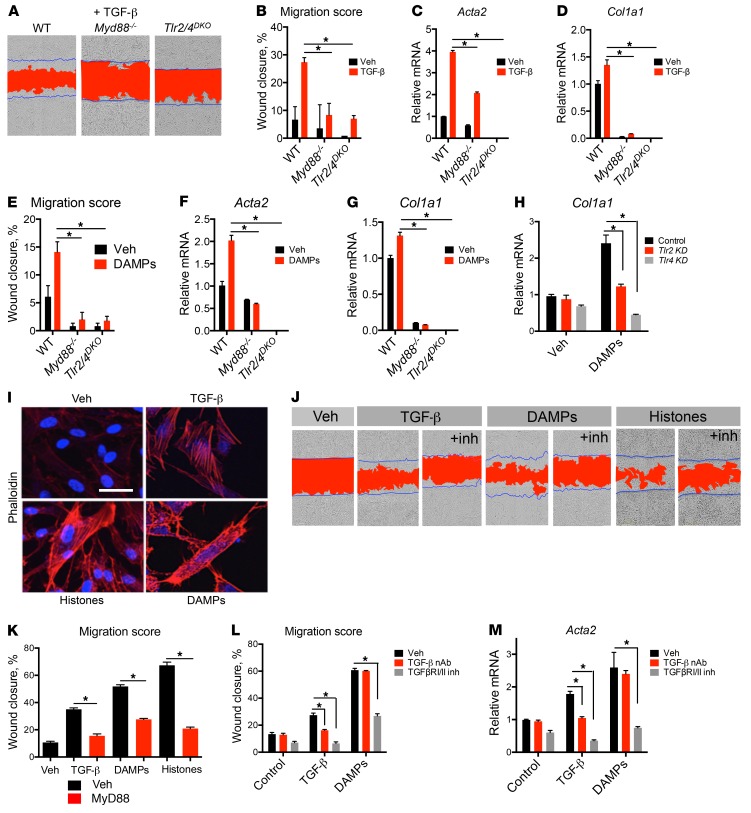
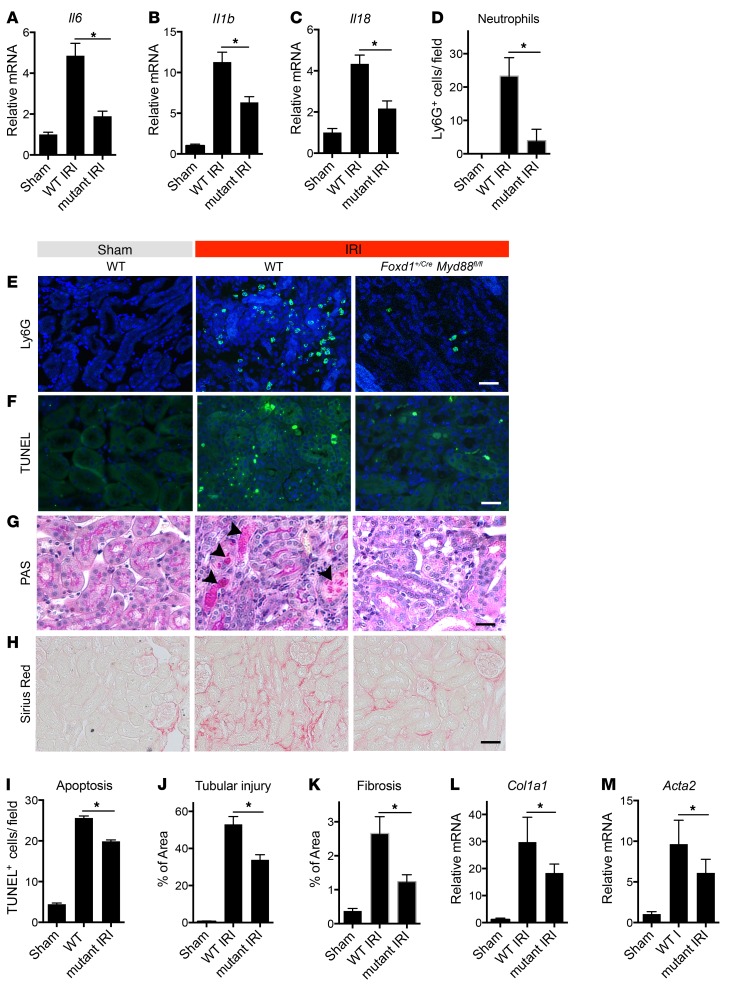
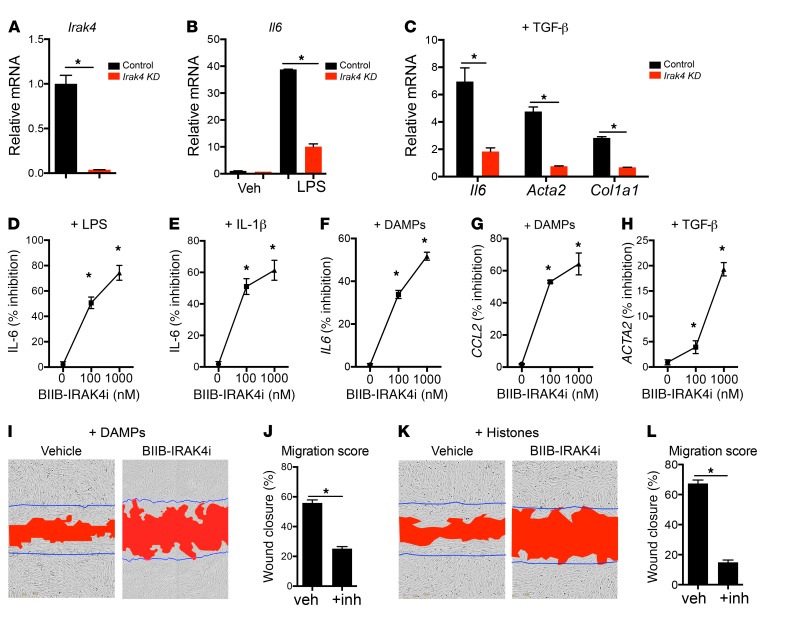
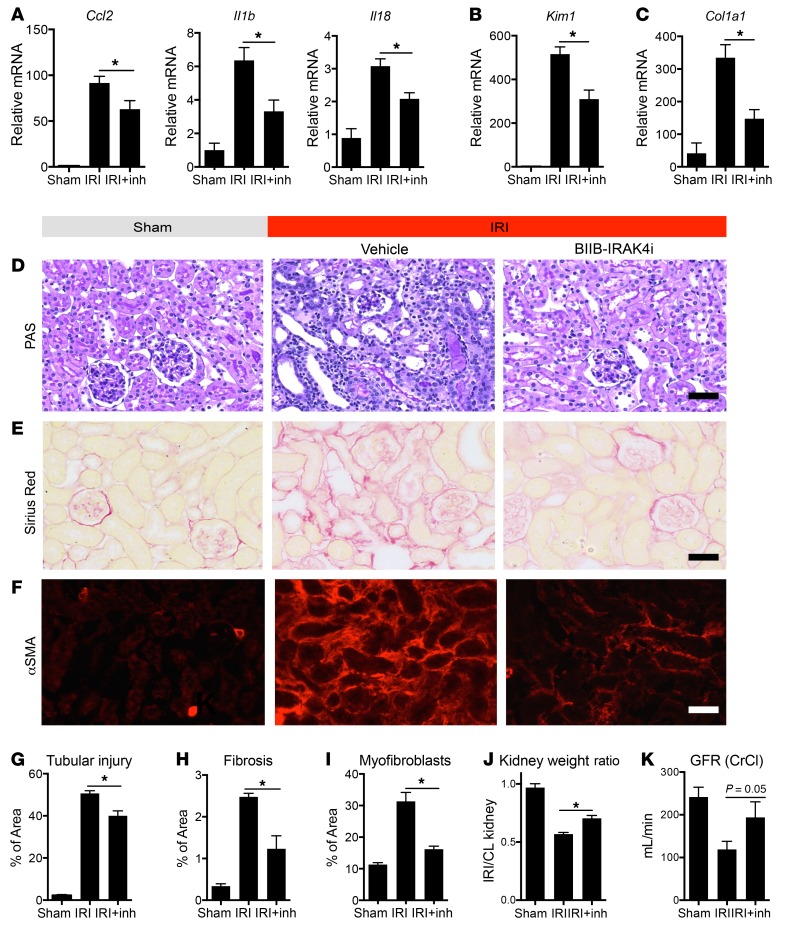
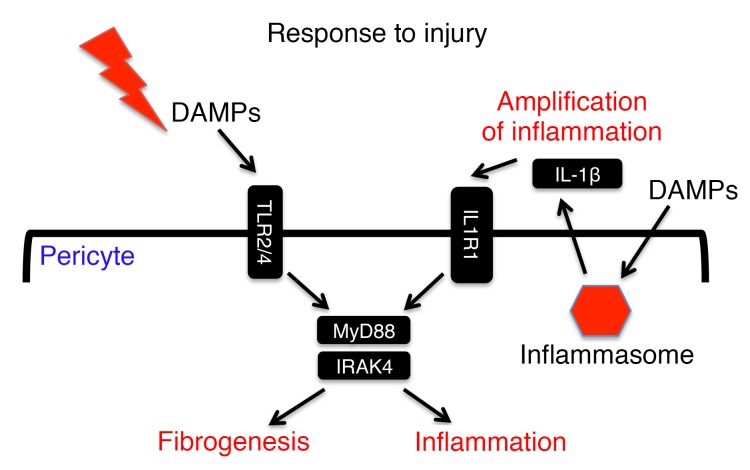
Fibrotic disease is associated with matrix deposition that results in the loss of organ function. Pericytes, the precursors of myofibroblasts, are a source of pathological matrix collagens and may be promising targets for treating fibrogenesis. Here, we have shown that pericytes activate a TLR2/4- and MyD88-dependent proinflammatory program in response to tissue injury. Similarly to classic immune cells, pericytes activate the NLRP3 inflammasome, leading to IL-1β and IL-18 secretion. Released IL-1β signals through pericyte MyD88 to amplify this response. Unexpectedly, we found that MyD88 and its downstream effector kinase IRAK4 intrinsically control pericyte migration and conversion to myofibroblasts. Specific ablation of MyD88 in pericytes or pharmacological inhibition of MyD88 signaling by an IRAK4 inhibitor in vivo protected against kidney injury by profoundly attenuating tissue injury, activation, and differentiation of myofibroblasts. Our data show that in pericytes, MyD88 and IRAK4 are key regulators of 2 major injury responses: inflammatory and fibrogenic. Moreover, these findings suggest that disruption of this MyD88-dependent pathway in pericytes might be a potential therapeutic approach to inhibit fibrogenesis and promote regeneration.
Conflict of interest statement
I.A. Leaf, B.G. Johnson, K.M. Guckian, I.G. Gomez, and J.S. Duffield own Biogen stock. K.M. Guckian, I.A. Leaf, and J.S. Duffield have filed a patent for the use of IRAK4 inhibition in the treatment of fibrosis (WO 2016011390 A1).
Figures
Comment in
-
Fibrosis: Pericyte MyD88 and IRAK4 signalling in fibrosis.Nat Rev Nephrol. 2017 Feb;13(2):63. doi: 10.1038/nrneph.2016.174. Epub 2016 Dec 5. Nat Rev Nephrol. 2017. PMID: 27916973 No abstract available.
References
-
- Zeisberg M, Kalluri R. Fibroblasts emerge via epithelial-mesenchymal transition in chronic kidney fibrosis. Front Biosci. 2008;13:6991–6998. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
