

Pulmonary endothelial activation caused by extracellular histones contributes to neutrophil activation in acute respiratory distress syndrome
- PMID: 27871277
- PMCID: PMC5117496
- DOI: 10.1186/s12931-016-0472-y
Pulmonary endothelial activation caused by extracellular histones contributes to neutrophil activation in acute respiratory distress syndrome
Abstract
Background: During the acute respiratory distress syndrome (ARDS), neutrophils play a central role in the pathogenesis, and their activation requires interaction with the endothelium. Extracellular histones have been recognized as pivotal inflammatory mediators. This study was to investigate the role of pulmonary endothelial activation during the extracellular histone-induced inflammatory response in ARDS.
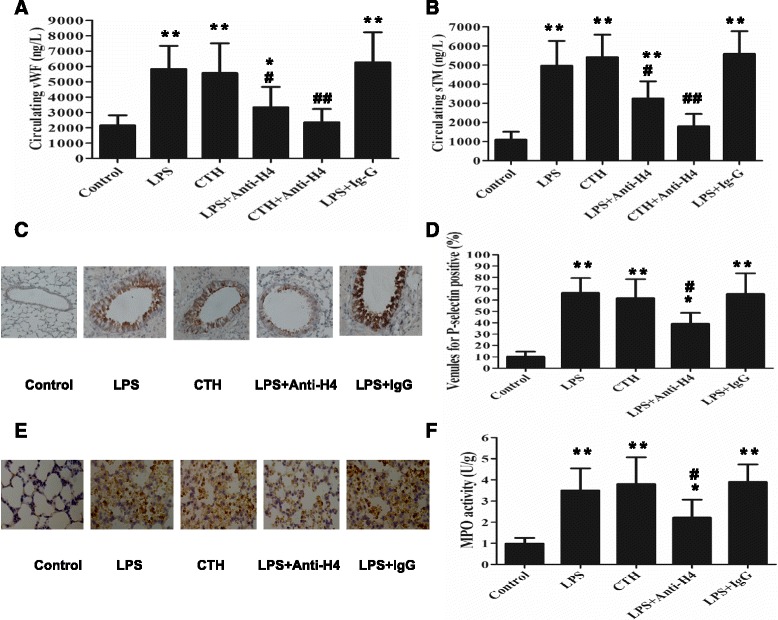
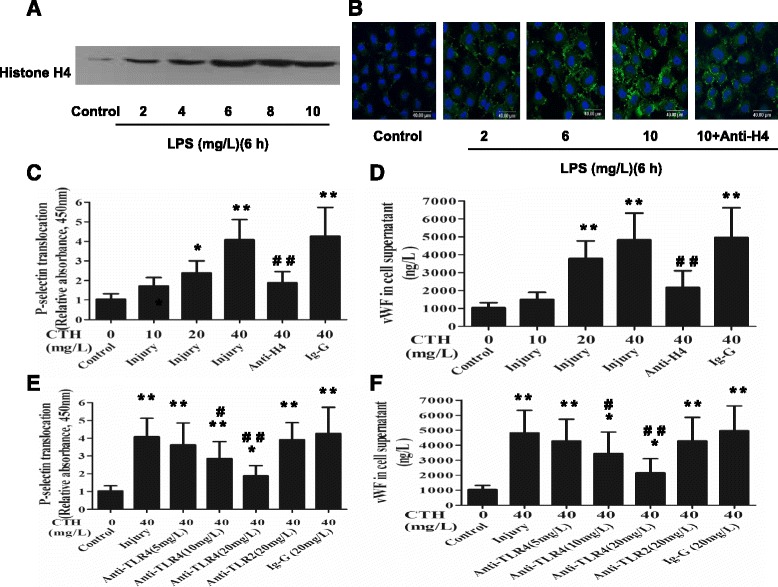
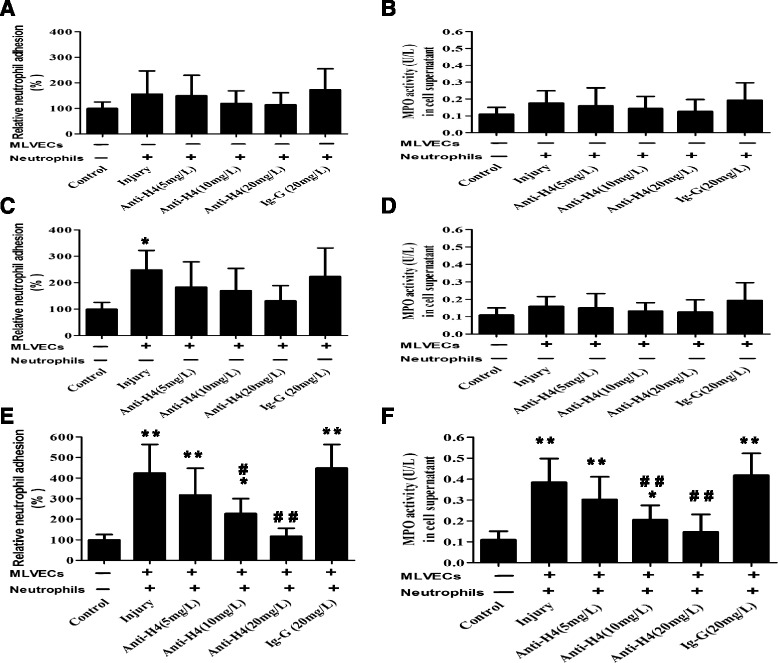
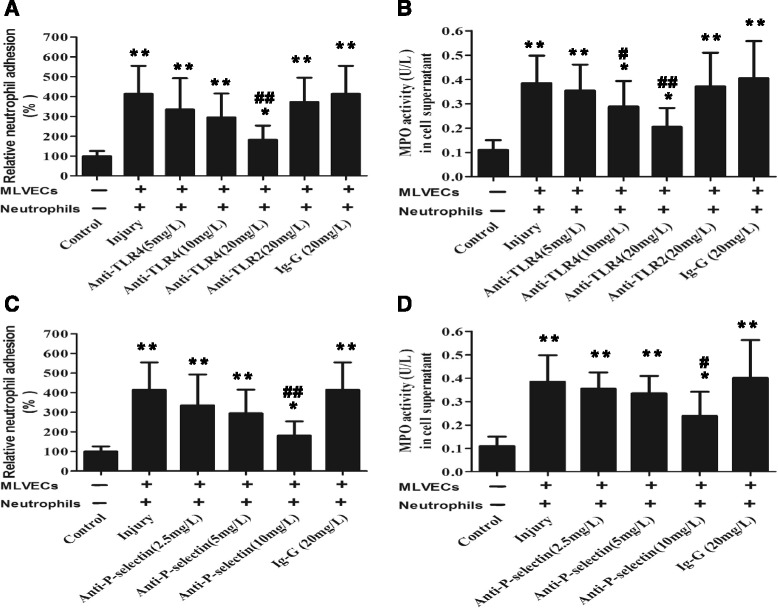
Methods: ARDS was induced in male C57BL/6 mice by intravenous injection with lipopolysaccharide (LPS) or exogenous histones. Concurrent with LPS administration, anti-histone H4 antibody (anti-H4) or non-specific IgG was administered to study the role of extracellular histones. The circulating von Willebrand factor (vWF) and soluble thrombomodulin (sTM) were measured with ELISA kits at the preset time points. Myeloperoxidase (MPO) activity in lung tissue was measured with a MPO detection kit. The translocation of P-selectin and neutrophil infiltration were measured by immunohistochemical detection. For in vitro studies, histone H4 in the supernatant of mouse lung vascular endothelial cells (MLVECs) was measured by Western blot. The binding of extracellular histones with endothelial membrane was examined by confocal laser microscopy. Endothelial P-selectin translocation was measured by cell surface ELISA. Adhesion of neutrophils to MLVECs was assessed with a color video digital camera.
Results: The results showed that during LPS-induced ARDS extracellular histones caused endothelial and neutrophil activation, as seen by P-selectin translocation, release of vWF, an increase of circulating sTM, lung neutrophil infiltration and increased MPO activity. Extracellular histones directly bound and activated MLVECs in a dose-dependent manner. On the contrary, the direct stimulatory effect of exogenous histones on neutrophils was very limited, as measured by neutrophil adhesion and MPO activity. With the contribution of activated endothelium, extracellular histones could effectively activating neutrophils. Both inhibiting the endothelial activation with an anti-toll like receptor (TLR) antibody and inhibiting the interaction of the endothelium with neutrophil using an anti-P-selectin antibody decreased the degree of neutrophil activation.
Conclusions: Extracellular histones are pro-inflammatory mediators in LPS-induced ARDS in mice. In addition to direct action to neutrophils, extracellular histones promote neutrophil adhesion and subsequent activation by first activating the pulmonary endothelium via TLR signaling. Thus, endothelial activation is important for extracellular histone-induced inflammatory injury.
Keywords: Acute respiratory distress syndrome; Endothelium; Extracellular histones; Inflammation; Neutrophil.
Figures
Similar articles
-
By activating endothelium histone H4 mediates oleic acid-induced acute respiratory distress syndrome.BMC Pulm Med. 2025 Jan 6;25(1):3. doi: 10.1186/s12890-024-03334-w. BMC Pulm Med. 2025. PMID: 39757148 Free PMC article.
-
[Activation of lung endothelial cells by extracellular histone in mice with acute respiratory distress syndrome].Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 2019 Oct 20;37(10):732-736. doi: 10.3760/cma.j.issn.1001-9391.2019.10.004. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 2019. PMID: 31726502 Chinese.
-
Adhesive Mechanisms of Histone-Induced Neutrophil-Endothelium Interactions in the Muscle Microcirculation.Eur Surg Res. 2016;56(1-2):19-31. doi: 10.1159/000441778. Epub 2015 Nov 18. Eur Surg Res. 2016. PMID: 26575178
-
The counter-intuitive role of the neutrophil in the acute respiratory distress syndrome.Br Med Bull. 2019 Sep 19;131(1):43-55. doi: 10.1093/bmb/ldz024. Br Med Bull. 2019. PMID: 31504234 Review.
-
Nanomedicine for acute respiratory distress syndrome: The latest application, targeting strategy, and rational design.Acta Pharm Sin B. 2021 Oct;11(10):3060-3091. doi: 10.1016/j.apsb.2021.04.023. Epub 2021 May 7. Acta Pharm Sin B. 2021. PMID: 33977080 Free PMC article. Review.
Cited by
-
Evolution of NETosis markers and DAMPs have prognostic value in critically ill COVID-19 patients.Sci Rep. 2021 Aug 3;11(1):15701. doi: 10.1038/s41598-021-95209-x. Sci Rep. 2021. PMID: 34344929 Free PMC article.
-
Hydrostatin-SN1, a Sea Snake-Derived Bioactive Peptide, Reduces Inflammation in a Mouse Model of Acute Lung Injury.Front Pharmacol. 2017 May 5;8:246. doi: 10.3389/fphar.2017.00246. eCollection 2017. Front Pharmacol. 2017. PMID: 28529485 Free PMC article.
-
Atherosclerosis in Rheumatoid Arthritis: Promoters and Opponents.Clin Rev Allergy Immunol. 2020 Feb;58(1):1-14. doi: 10.1007/s12016-018-8714-z. Clin Rev Allergy Immunol. 2020. PMID: 30259381 Review.
-
Biomarkers in Community-Acquired Pneumonia (Cardiac and Non-Cardiac).J Clin Med. 2020 Feb 18;9(2):549. doi: 10.3390/jcm9020549. J Clin Med. 2020. PMID: 32085380 Free PMC article. Review.
-
Endothelial cell dynamics in sepsis-induced acute lung injury and acute respiratory distress syndrome: pathogenesis and therapeutic implications.Cell Commun Signal. 2024 Apr 25;22(1):241. doi: 10.1186/s12964-024-01620-y. Cell Commun Signal. 2024. PMID: 38664775 Free PMC article. Review.
References
-
- Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, et al. Acute respiratory distress syndrome: the Berlin definition. JAMA. 2012;307:2526–33. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
