

Autolysosome biogenesis and developmental senescence are regulated by both Spns1 and v-ATPase
- PMID: 27875093
- PMCID: PMC5324850
- DOI: 10.1080/15548627.2016.1256934
Autolysosome biogenesis and developmental senescence are regulated by both Spns1 and v-ATPase
Abstract
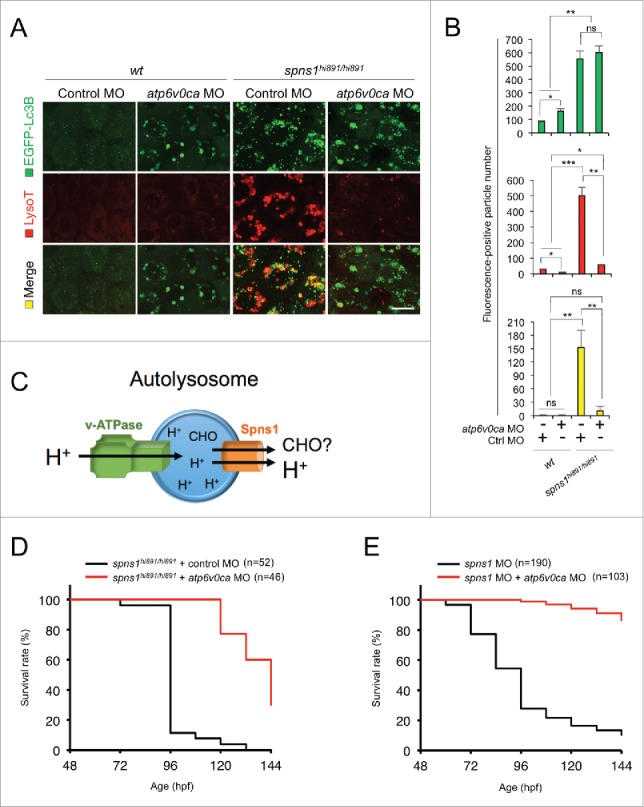
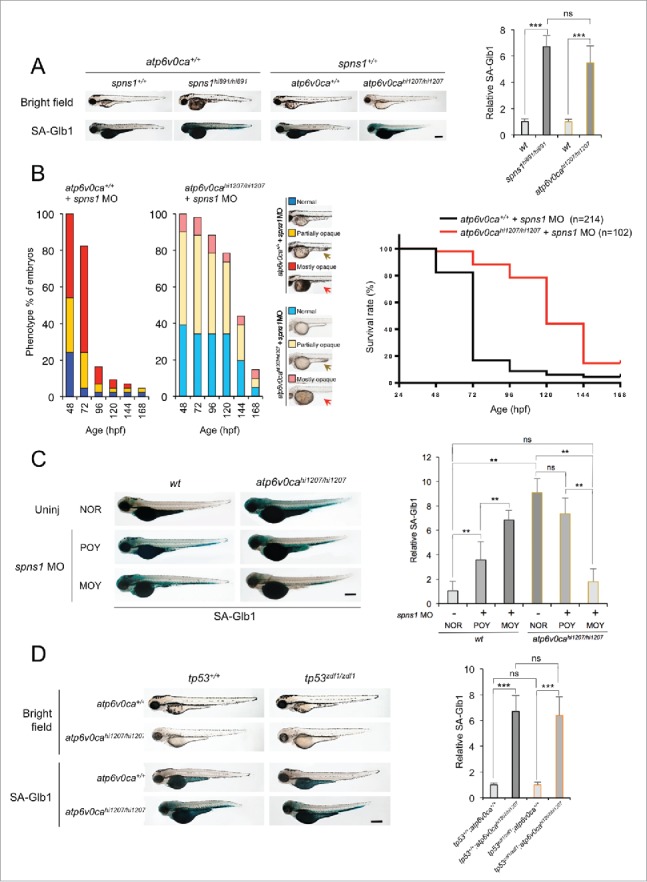
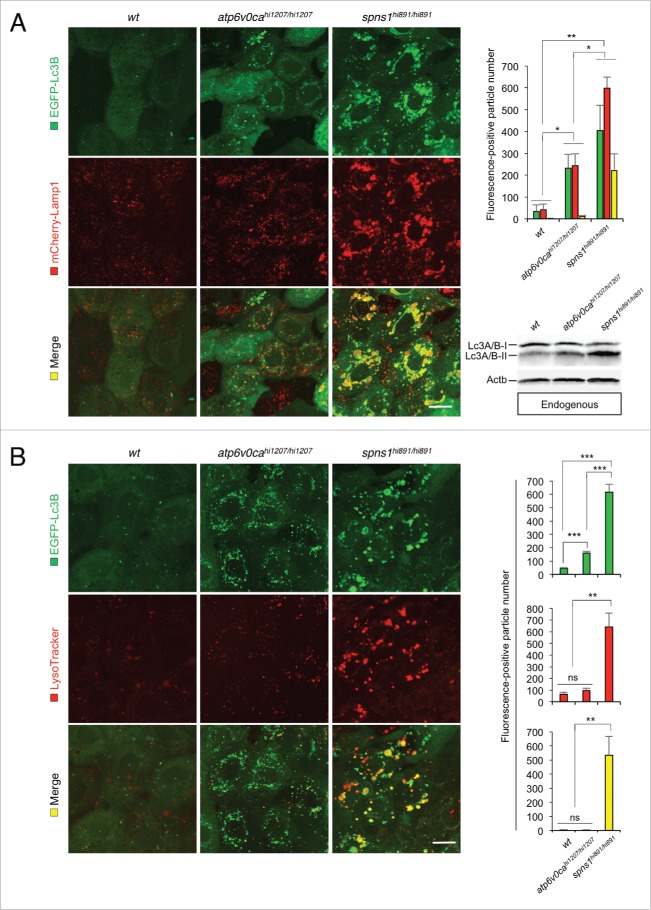
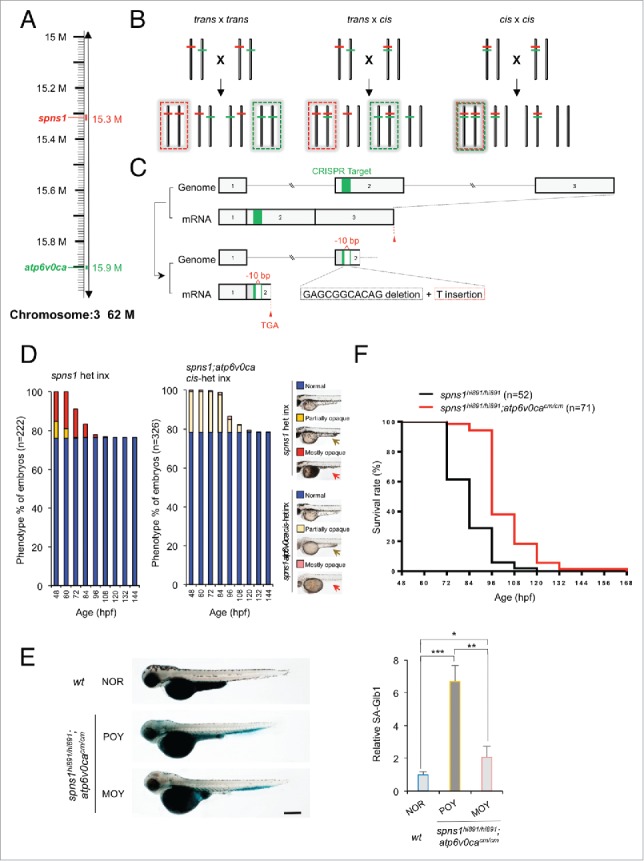
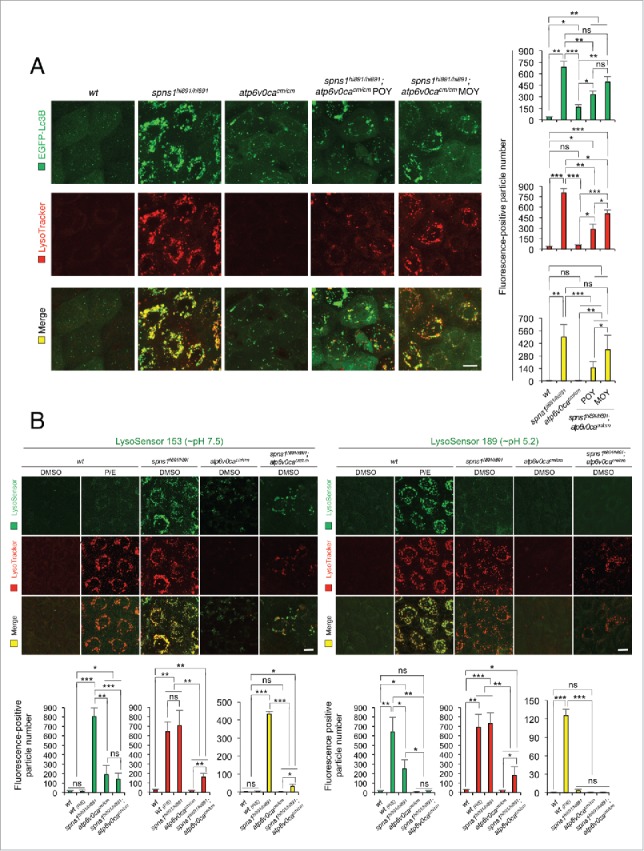
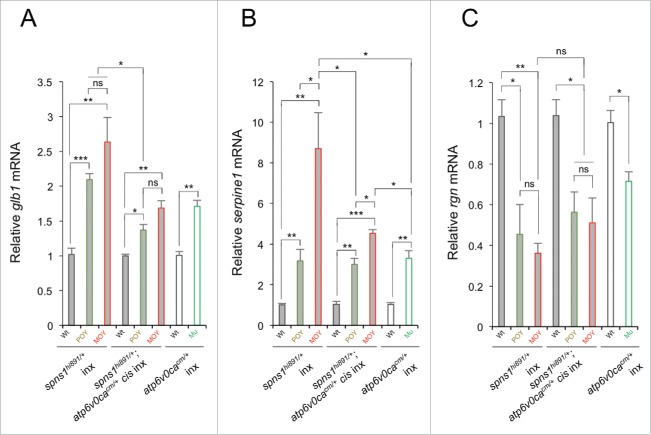
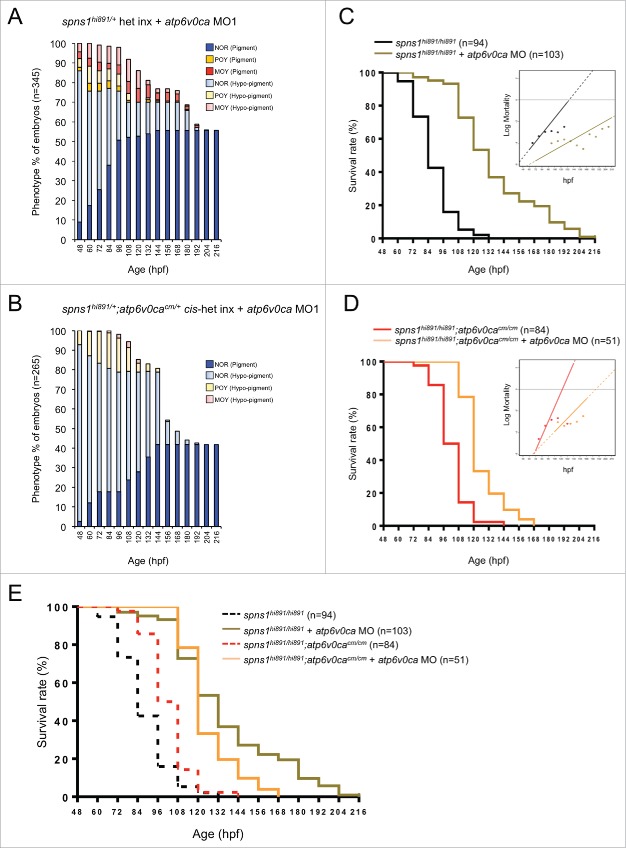
Spns1 (Spinster homolog 1 [Drosophila]) in vertebrates, as well as Spin (Spinster) in Drosophila, is a hypothetical lysosomal H+-carbohydrate transporter, which functions at a late stage of macroautophagy (hereafter autophagy). The Spin/Spns1 defect induces aberrant autolysosome formation that leads to developmental senescence in the embryonic stage and premature aging symptoms in adulthood. However, the molecular mechanism by which loss of Spin/Spns1 leads to the specific pathogenesis remains to be elucidated. Using chemical, genetic and CRISPR/Cas9-mediated genome-editing approaches in zebrafish, we investigated and determined a mechanism that suppresses embryonic senescence as well as autolysosomal impairment mediated by Spns1 deficiency. Unexpectedly, we found that a concurrent disruption of the vacuolar-type H+-ATPase (v-ATPase) subunit gene, atp6v0ca (ATPase, H+ transporting, lysosomal, V0 subunit ca) led to suppression of the senescence induced by the Spns1 defect, whereas the sole loss of Atp6v0ca led to senescent embryos similar to the single spns1 mutation. Moreover, we discovered that the combined stable defect seen in the presence of both the spns1 and atp6v0ca mutant genes still subsequently induced premature autophagosome-lysosome fusion marked by insufficient acidity, while extending developmental life span, compared with the solely mutated spns1 defect. Our data suggest that Spns1 and the v-ATPase orchestrate proper autolysosomal biogenesis with optimal acidification that is critically linked to developmental senescence and survival.
Keywords: aging; autophagy; lysosome; senescence; spinster; vacuolar-type H+-ATPase; zebrafish.
Figures
References
-
- Cao L, Li W, Kim S, Brodie SG, Deng CX. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev 2003; 17(2):201; PMID:12533509; http://dx.doi.org/ 10.1101/gad.1050003 - DOI - PMC - PubMed
-
- Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, Wu JJ, Peng TN, Chen J, Nussenzweig A, et al.. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell 2009; 35(4):534; PMID:19716796; http://dx.doi.org/ 10.1016/j.molcel.2009.06.037 - DOI - PMC - PubMed
-
- Kishi S, Bayliss PE, Uchiyama J, Koshimizu E, Qi J, Nanjappa P, Imamura S, Islam A, Neuberg D, Amsterdam A, et al.. The identification of zebrafish mutants showing alterations in senescence-associated biomarkers. PLoS Genet 2008; 4(8):e1000152; PMID:18704191; http://dx.doi.org/ 10.1371/journal.pgen.1000152 - DOI - PMC - PubMed
-
- Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, et al.. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013; 155(5):1119; PMID:24238961; http://dx.doi.org/ 10.1016/j.cell.2013.10.041 - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials