

Attenuation of Blood-Brain Barrier Breakdown and Hyperpermeability by Calpain Inhibition
- PMID: 27875293
- PMCID: PMC5207131
- DOI: 10.1074/jbc.M116.735365
Attenuation of Blood-Brain Barrier Breakdown and Hyperpermeability by Calpain Inhibition
Abstract
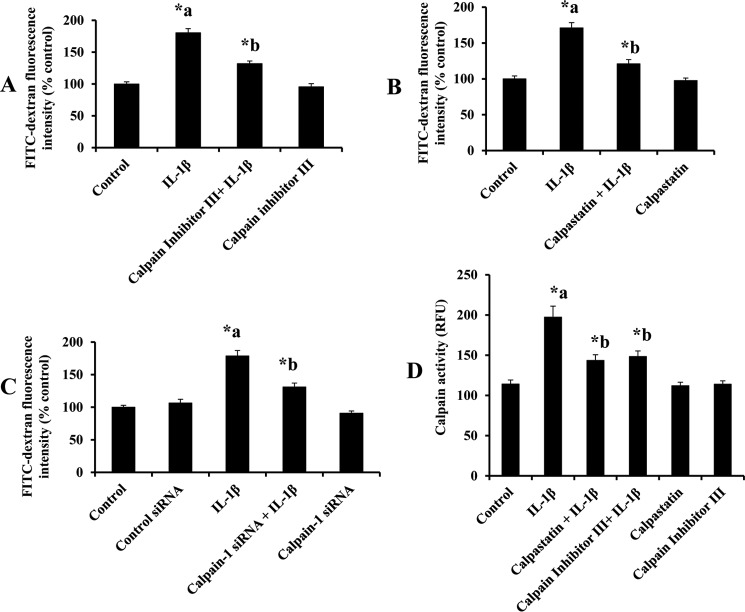
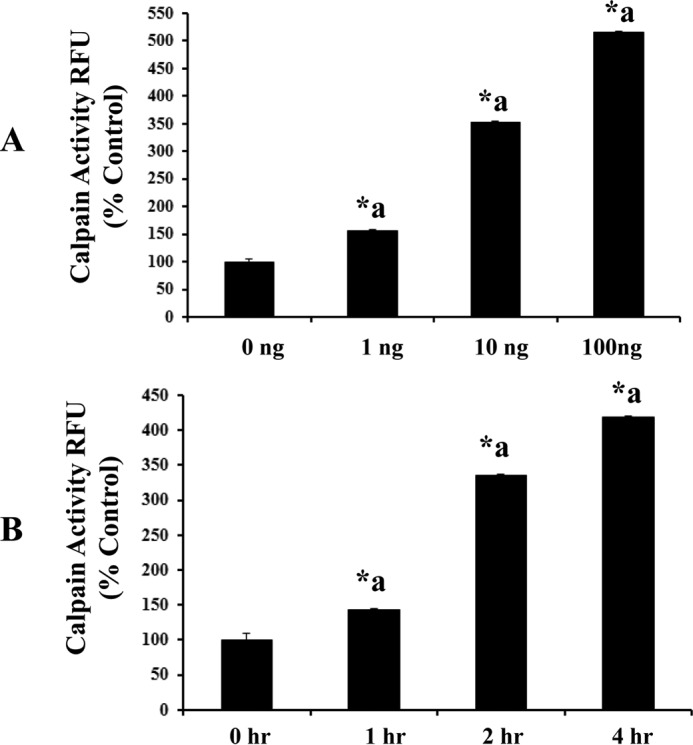
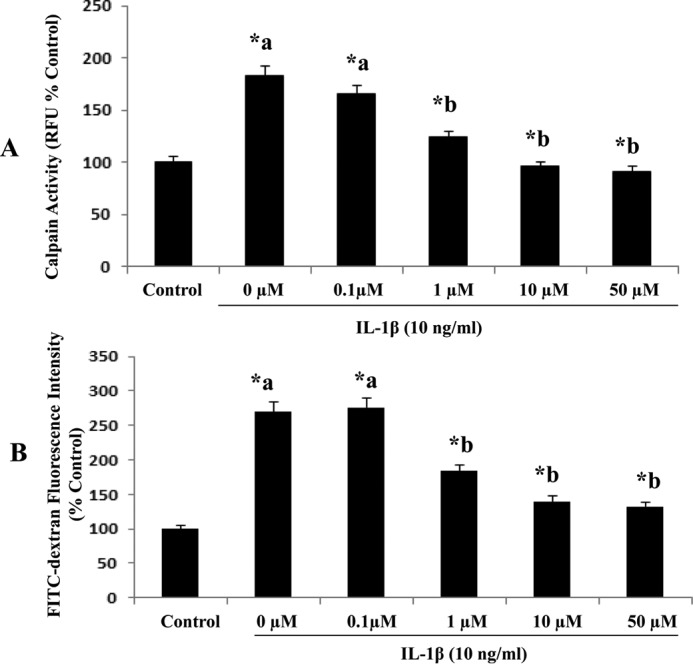
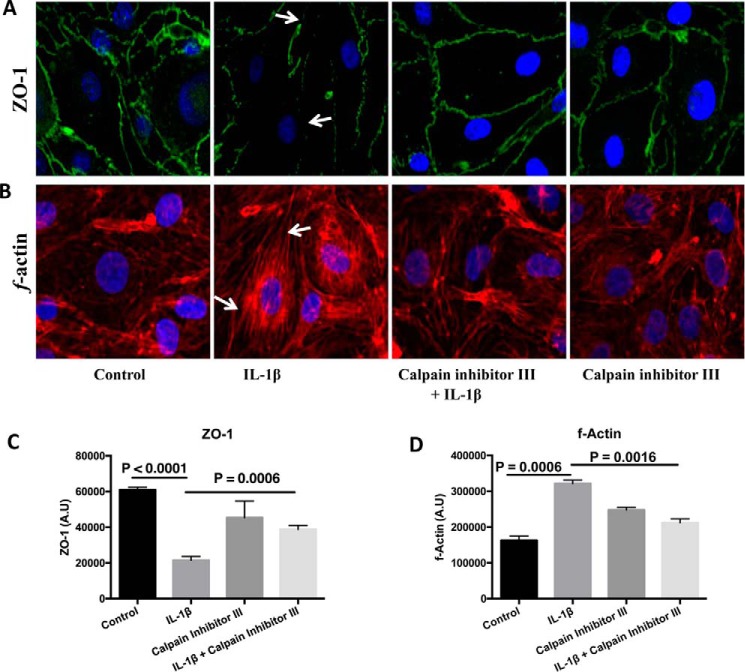
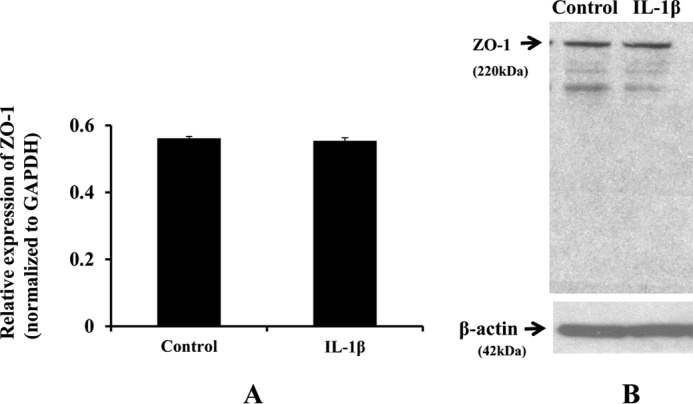
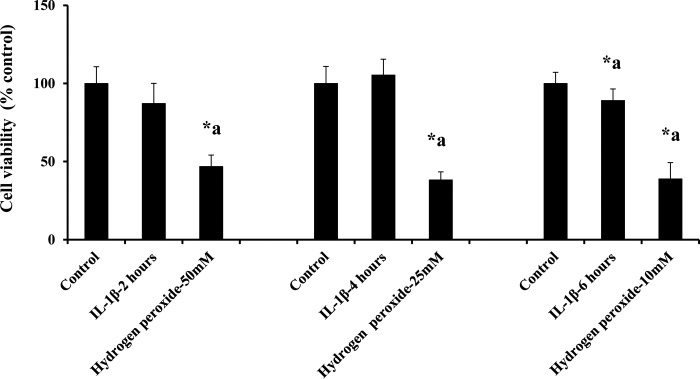
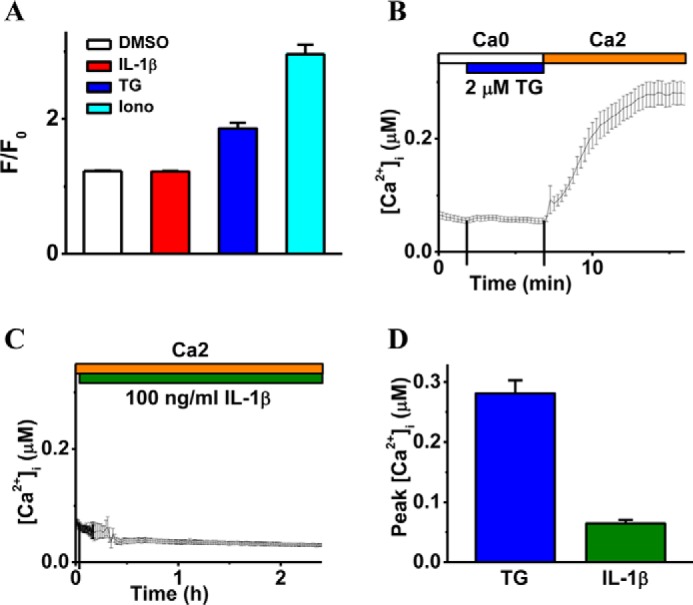
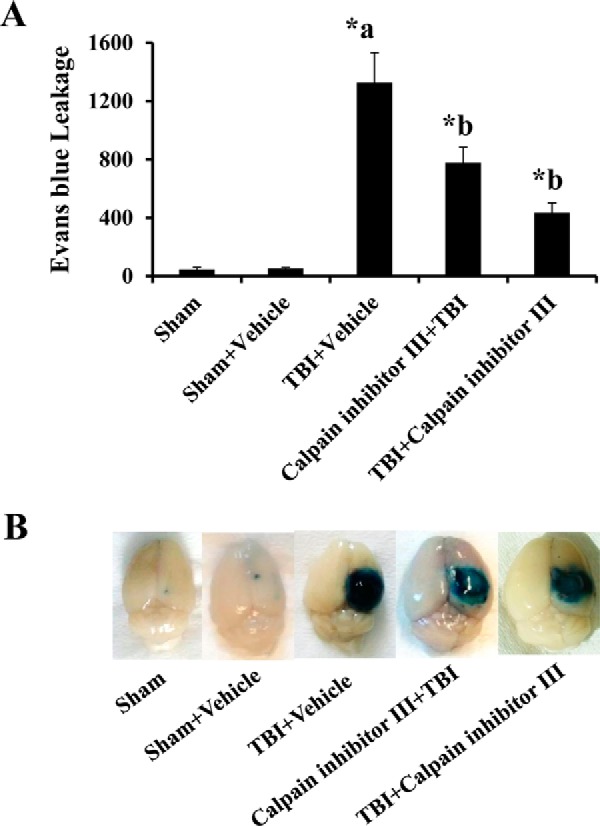
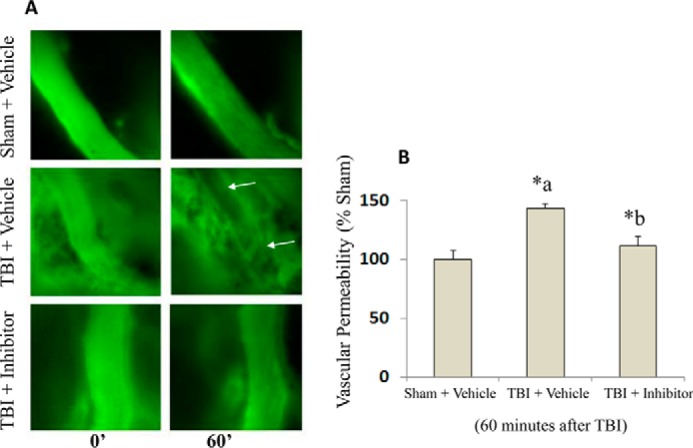
Blood-brain barrier (BBB) breakdown and the associated microvascular hyperpermeability followed by brain edema are hallmark features of several brain pathologies, including traumatic brain injuries (TBI). Recent studies indicate that pro-inflammatory cytokine interleukin-1β (IL-1β) that is up-regulated following traumatic injuries also promotes BBB dysfunction and hyperpermeability, but the underlying mechanisms are not clearly known. The objective of this study was to determine the role of calpains in mediating BBB dysfunction and hyperpermeability and to test the effect of calpain inhibition on the BBB following traumatic insults to the brain. In these studies, rat brain microvascular endothelial cell monolayers exposed to calpain inhibitors (calpain inhibitor III and calpastatin) or transfected with calpain-1 siRNA demonstrated attenuation of IL-1β-induced monolayer hyperpermeability. Calpain inhibition led to protection against IL-1β-induced loss of zonula occludens-1 (ZO-1) at the tight junctions and alterations in F-actin cytoskeletal assembly. IL-1β treatment had no effect on ZO-1 gene (tjp1) or protein expression. Calpain inhibition via calpain inhibitor III and calpastatin decreased IL-1β-induced calpain activity significantly (p < 0.05). IL-1β had no detectable effect on intracellular calcium mobilization or endothelial cell viability. Furthermore, calpain inhibition preserved BBB integrity/permeability in a mouse controlled cortical impact model of TBI when studied using Evans blue assay and intravital microscopy. These studies demonstrate that calpain-1 acts as a mediator of IL-1β-induced loss of BBB integrity and permeability by altering tight junction integrity, promoting the displacement of ZO-1, and disorganization of cytoskeletal assembly. IL-1β-mediated alterations in permeability are neither due to the changes in ZO-1 expression nor cell viability. Calpain inhibition has beneficial effects against TBI-induced BBB hyperpermeability.
Keywords: calpain; inflammation; inhibitor; interleukin 1 (IL-1); vascular biology.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Alluri H., Wiggins-Dohlvik K., Davis M. L., Huang J. H., and Tharakan B. (2015) Blood-brain barrier dysfunction following traumatic brain injury. Metab. Brain Dis. 30, 1093–1104 - PubMed
-
- Mayhan W. (2001) Regulation of blood–brain barrier permeability. Microcirculation 8, 1108–1110 - PubMed
-
- Blamire A. M., Anthony D. C., Rajagopalan B., Sibson N. R., Perry V. H., and Styles P. (2000) Interleukin-1β-induced changes in blood-brain barrier permeability, apparent diffusion coefficient, and cerebral blood volume in the rat brain: a magnetic resonance study. J. Neurosci. 20, 8153–8159 - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
