

Extending Halogen-based Medicinal Chemistry to Proteins: IODO-INSULIN AS A CASE STUDY
- PMID: 27875310
- PMCID: PMC5207135
- DOI: 10.1074/jbc.M116.761015
Extending Halogen-based Medicinal Chemistry to Proteins: IODO-INSULIN AS A CASE STUDY
Abstract
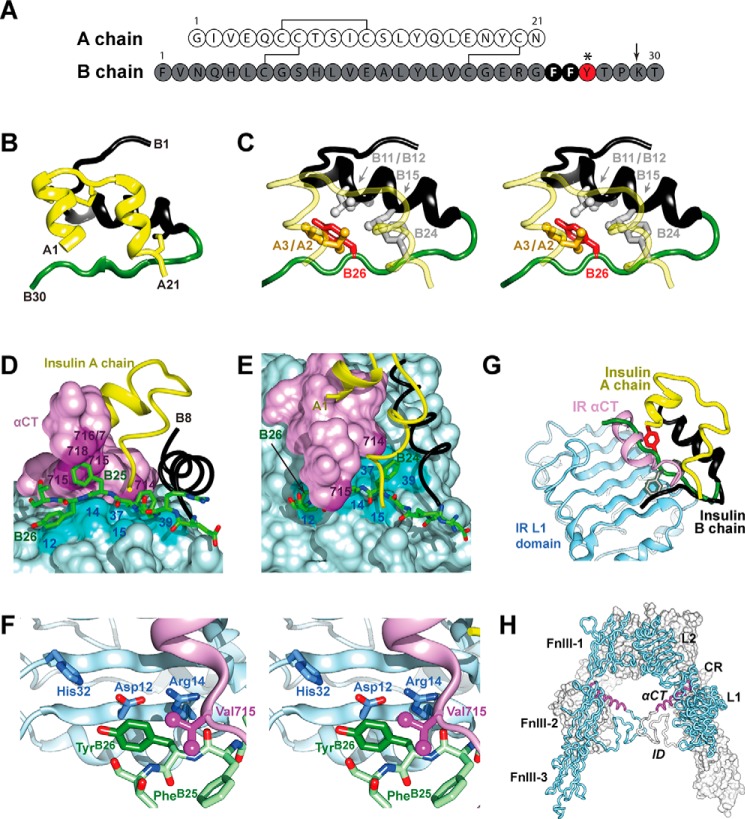
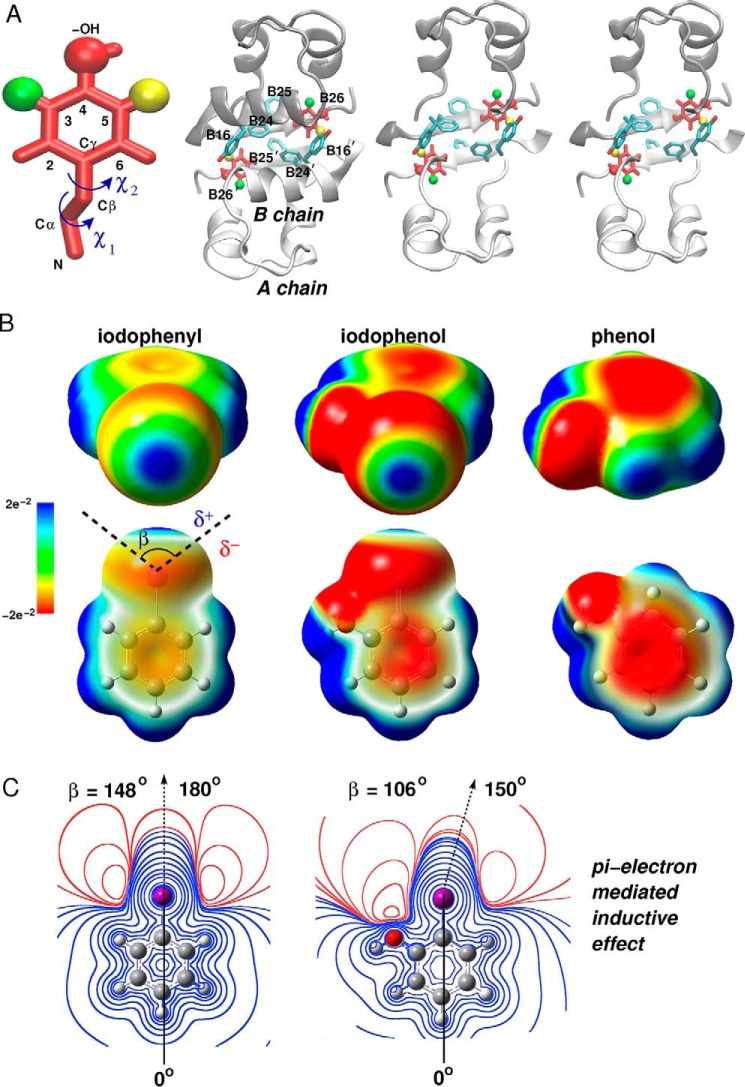
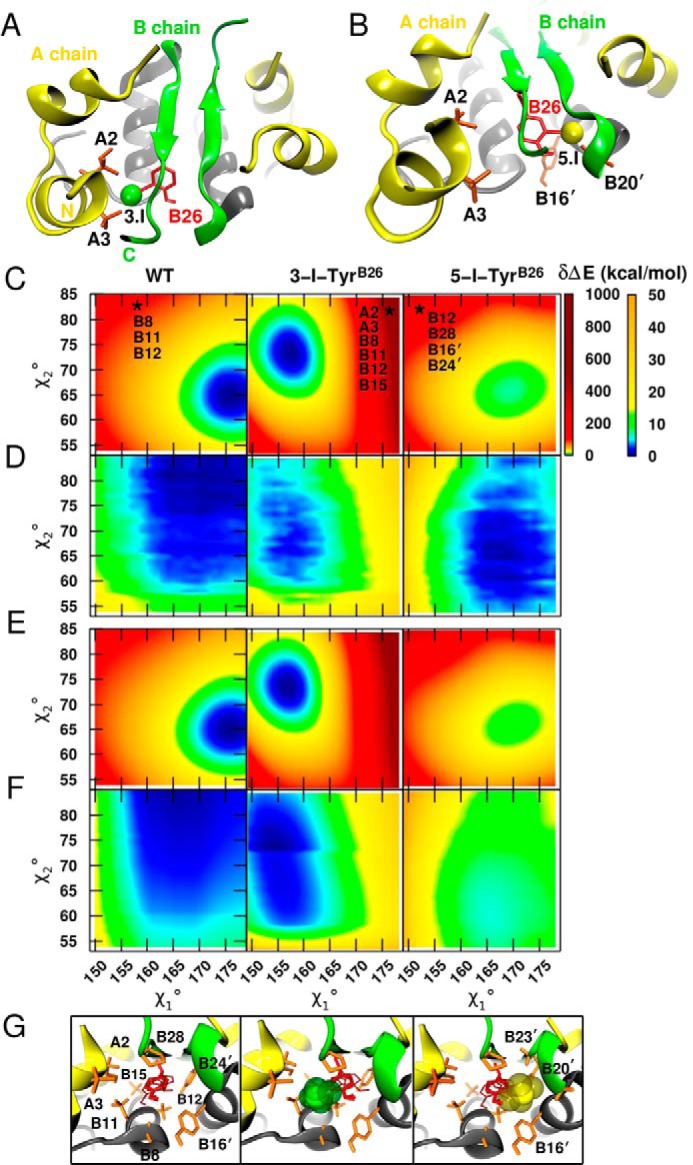
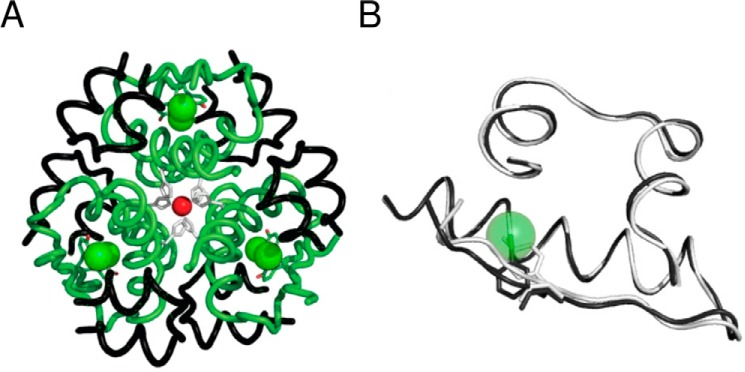
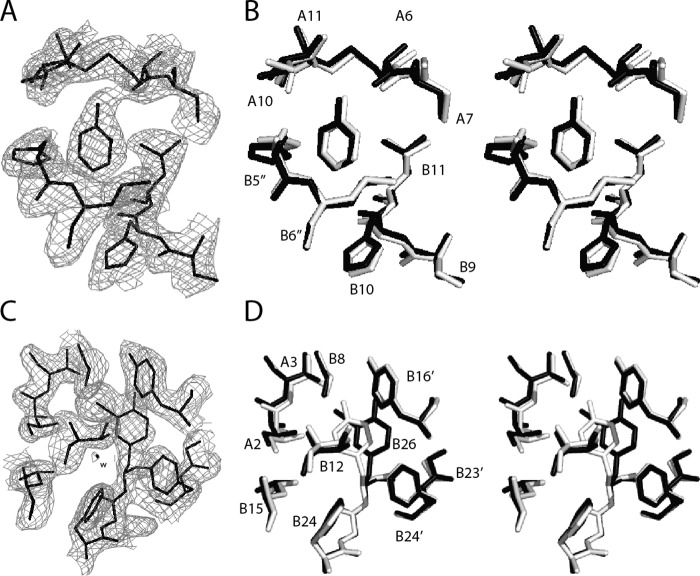
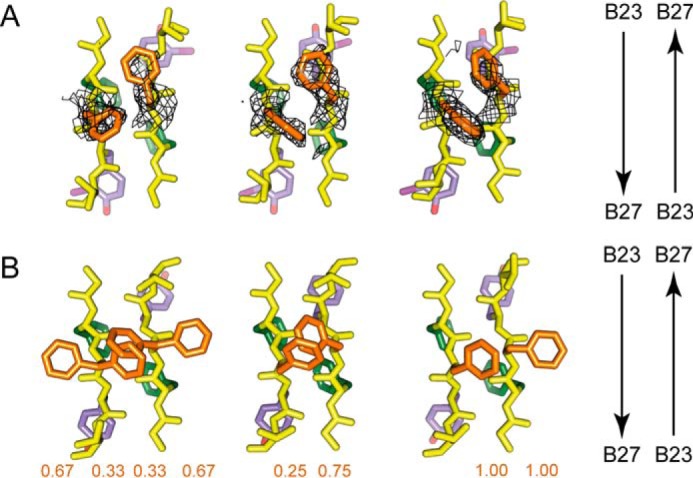
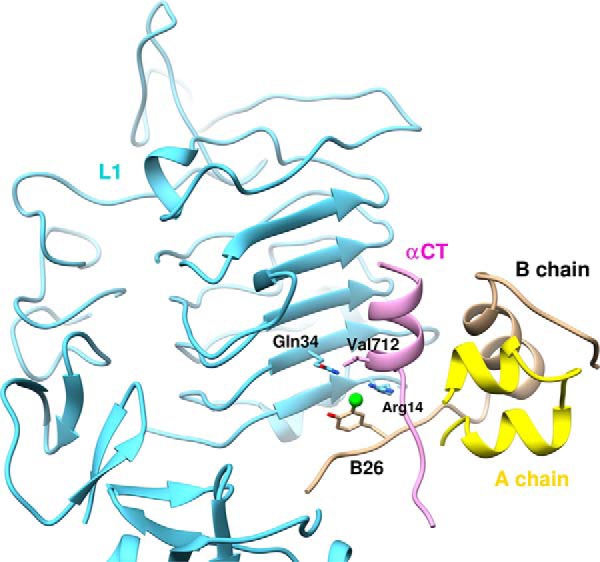
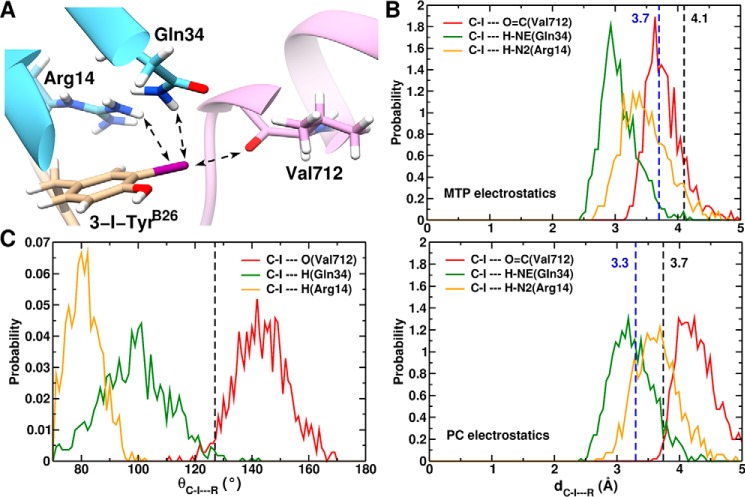
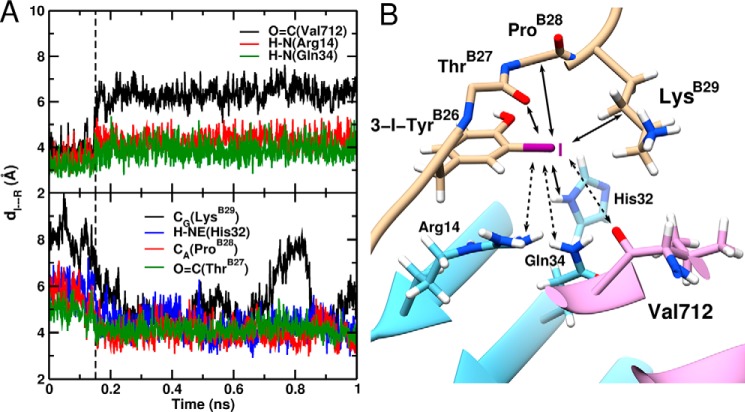
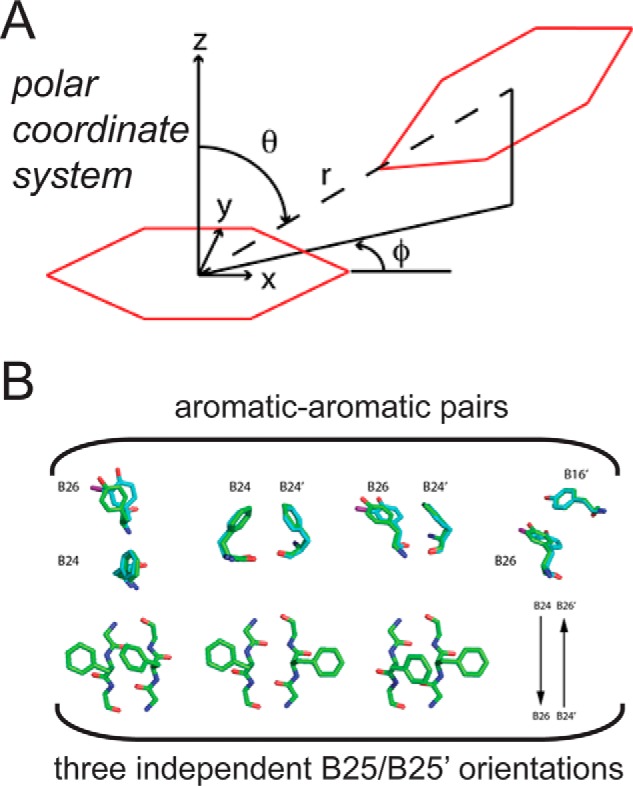
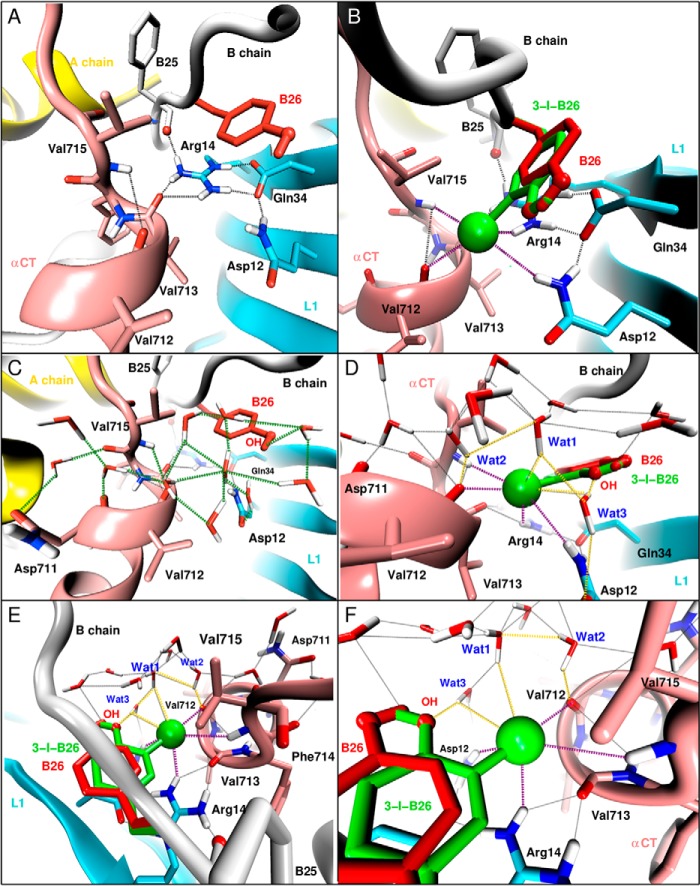
Insulin, a protein critical for metabolic homeostasis, provides a classical model for protein design with application to human health. Recent efforts to improve its pharmaceutical formulation demonstrated that iodination of a conserved tyrosine (TyrB26) enhances key properties of a rapid-acting clinical analog. Moreover, the broad utility of halogens in medicinal chemistry has motivated the use of hybrid quantum- and molecular-mechanical methods to study proteins. Here, we (i) undertook quantitative atomistic simulations of 3-[iodo-TyrB26]insulin to predict its structural features, and (ii) tested these predictions by X-ray crystallography. Using an electrostatic model of the modified aromatic ring based on quantum chemistry, the calculations suggested that the analog, as a dimer and hexamer, exhibits subtle differences in aromatic-aromatic interactions at the dimer interface. Aromatic rings (TyrB16, PheB24, PheB25, 3-I-TyrB26, and their symmetry-related mates) at this interface adjust to enable packing of the hydrophobic iodine atoms within the core of each monomer. Strikingly, these features were observed in the crystal structure of a 3-[iodo-TyrB26]insulin analog (determined as an R6 zinc hexamer). Given that residues B24-B30 detach from the core on receptor binding, the environment of 3-I-TyrB26 in a receptor complex must differ from that in the free hormone. Based on the recent structure of a "micro-receptor" complex, we predict that 3-I-TyrB26 engages the receptor via directional halogen bonding and halogen-directed hydrogen bonding as follows: favorable electrostatic interactions exploiting, respectively, the halogen's electron-deficient σ-hole and electronegative equatorial band. Inspired by quantum chemistry and molecular dynamics, such "halogen engineering" promises to extend principles of medicinal chemistry to proteins.
Keywords: diabetes; diabetes mellitus; hormone; molecular dynamics; non-standard mutagenesis; quantum chemistry; quantum mechanics; weakly polar.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Dodson G., and Steiner D. (1998) The role of assembly in insulin's biosynthesis. Curr. Opin. Struct. Biol. 8, 189–194 - PubMed
-
- Baker E. N., Blundell T. L., Cutfield J. F., Cutfield S. M., Dodson E. J., Dodson G. G., Hodgkin D. M., Hubbard R. E., Isaacs N. W., and Reynolds C. D. (1988) The structure of 2Zn pig insulin crystals at 1.5 Å resolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 319, 369–456 - PubMed
-
- Brange J., Ribel U., Hansen J. F., Dodson G., Hansen M. T., Havelund S., Melberg S. G., Norris F., Norris K., and Snel L. (1988) Monomeric insulins obtained by protein engineering and their medical implications. Nature 333, 679–682 - PubMed
-
- Inouye K., Watanabe K., Tochino Y., Kobayashi M., and Shigeta Y. (1981) Semisynthesis and properties of some insulin analogs. Biopolymers 20, 1845–1858 - PubMed
-
- Pandyarajan V., Smith B. J., Phillips N. B., Whittaker L., Cox G. P., Wickramasinghe N., Menting J. G., Wan Z.-L., Whittaker J., Ismail-Beigi F., Lawrence M. C., and Weiss M. A. (2014) Aromatic anchor at an invariant hormone-receptor interface function of insulin residue B24 with application to protein design. J. Biol. Chem. 289, 34709–34727 - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
