

Alignment-free Transcriptomic and Metatranscriptomic Comparison Using Sequencing Signatures with Variable Length Markov Chains
- PMID: 27876823
- PMCID: PMC5120338
- DOI: 10.1038/srep37243
Alignment-free Transcriptomic and Metatranscriptomic Comparison Using Sequencing Signatures with Variable Length Markov Chains
Abstract
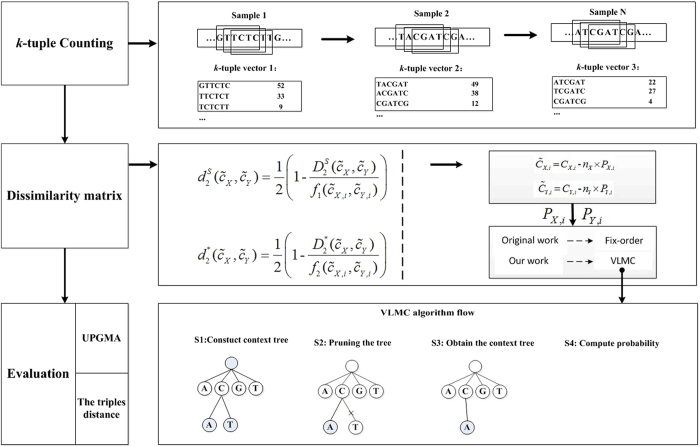
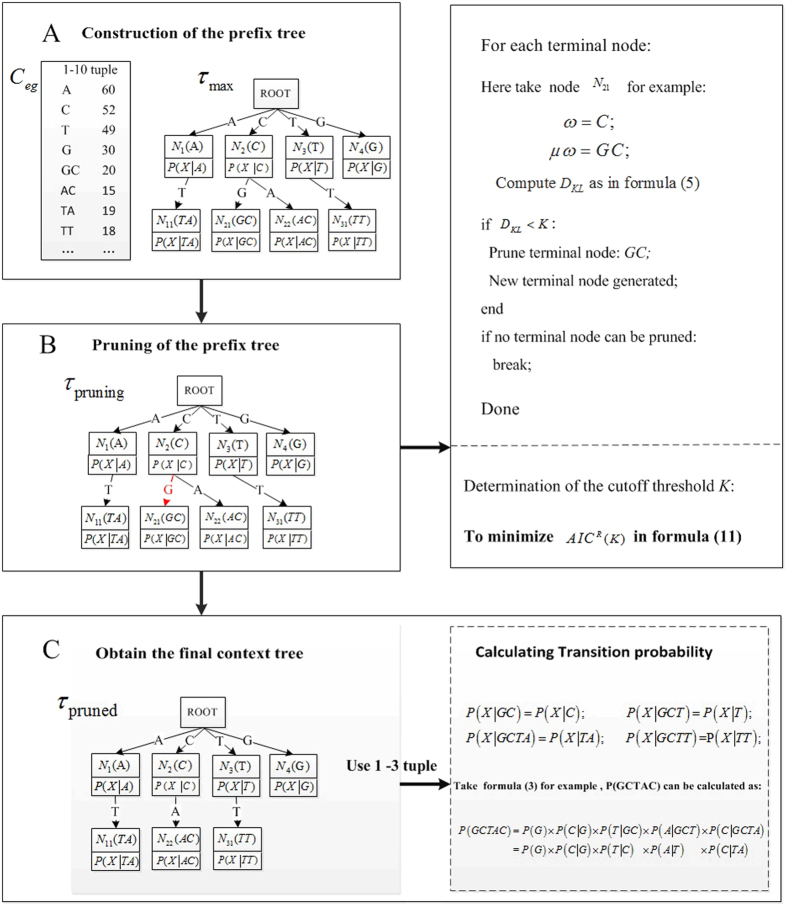
The comparison between microbial sequencing data is critical to understand the dynamics of microbial communities. The alignment-based tools analyzing metagenomic datasets require reference sequences and read alignments. The available alignment-free dissimilarity approaches model the background sequences with Fixed Order Markov Chain (FOMC) yielding promising results for the comparison of microbial communities. However, in FOMC, the number of parameters grows exponentially with the increase of the order of Markov Chain (MC). Under a fixed high order of MC, the parameters might not be accurately estimated owing to the limitation of sequencing depth. In our study, we investigate an alternative to FOMC to model background sequences with the data-driven Variable Length Markov Chain (VLMC) in metatranscriptomic data. The VLMC originally designed for long sequences was extended to apply to high-throughput sequencing reads and the strategies to estimate the corresponding parameters were developed. The flexible number of parameters in VLMC avoids estimating the vast number of parameters of high-order MC under limited sequencing depth. Different from the manual selection in FOMC, VLMC determines the MC order adaptively. Several beta diversity measures based on VLMC were applied to compare the bacterial RNA-Seq and metatranscriptomic datasets. Experiments show that VLMC outperforms FOMC to model the background sequences in transcriptomic and metatranscriptomic samples. A software pipeline is available at https://d2vlmc.codeplex.com.
Figures
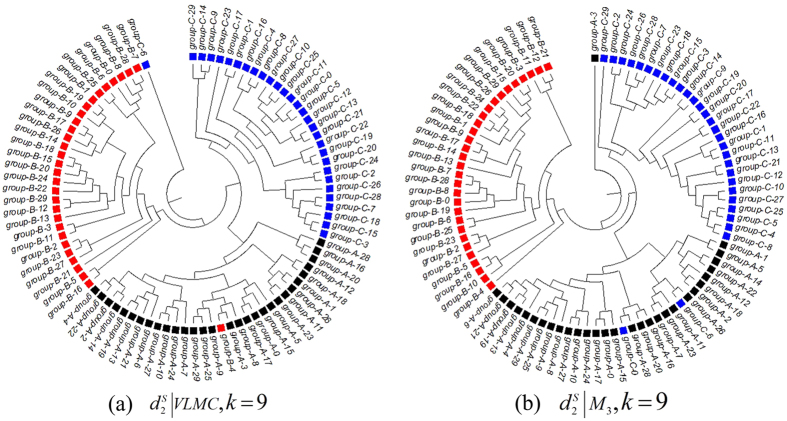
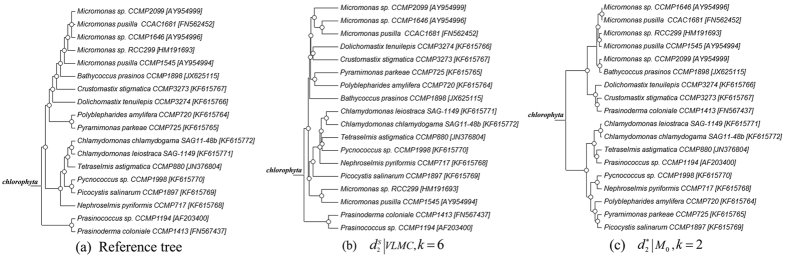
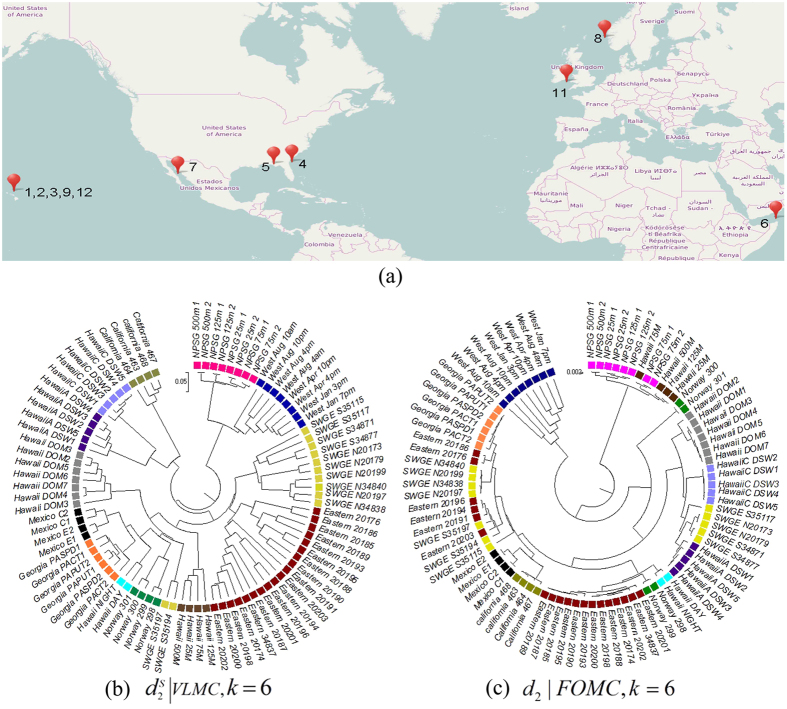
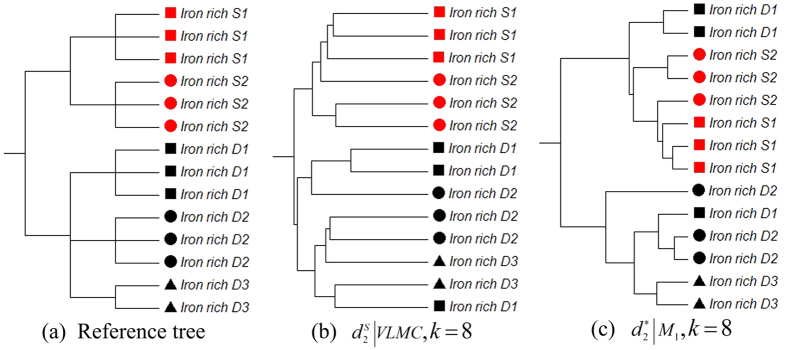
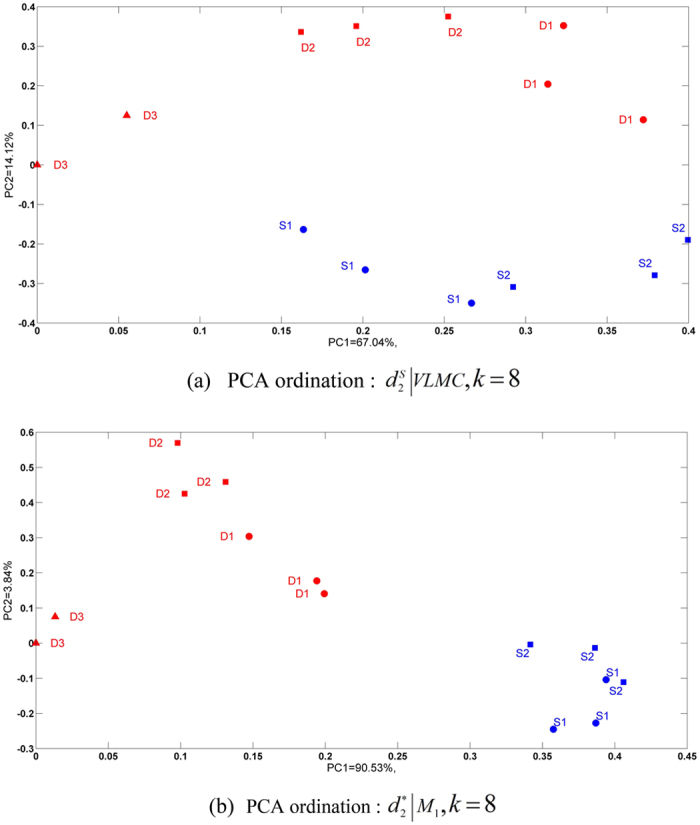
 and k = 6. (c) The clustering tree with FOMC using
and k = 6. (c) The clustering tree with FOMC using  and k = 6. *‘SWGE’ (Dataset 10 in Supplemental Table S4 in section 2) samples were collected from different locations with two research cruises in the Equatorial North Atlantic Ocean and South Pacific Subtropical gyre.
and k = 6. *‘SWGE’ (Dataset 10 in Supplemental Table S4 in section 2) samples were collected from different locations with two research cruises in the Equatorial North Atlantic Ocean and South Pacific Subtropical gyre.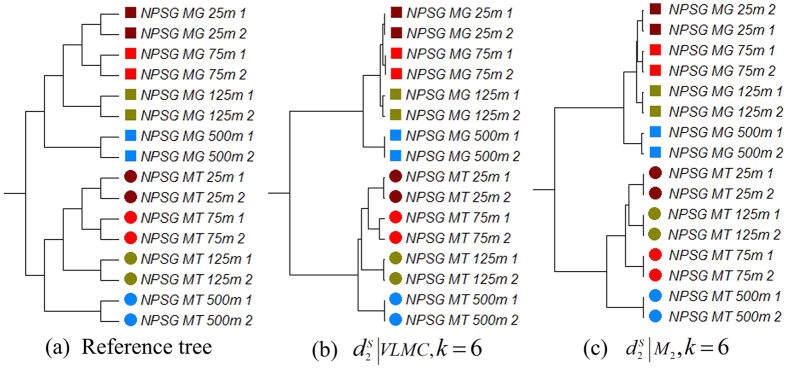
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
