

Altered Smooth Muscle Cell Force Generation as a Driver of Thoracic Aortic Aneurysms and Dissections
- PMID: 27879251
- PMCID: PMC5222685
- DOI: 10.1161/ATVBAHA.116.303229
Altered Smooth Muscle Cell Force Generation as a Driver of Thoracic Aortic Aneurysms and Dissections
Erratum in
-
Correction.Arterioscler Thromb Vasc Biol. 2017 Feb;37(2):e12. doi: 10.1161/ATV.0000000000000045. Arterioscler Thromb Vasc Biol. 2017. PMID: 28122778 No abstract available.
Abstract
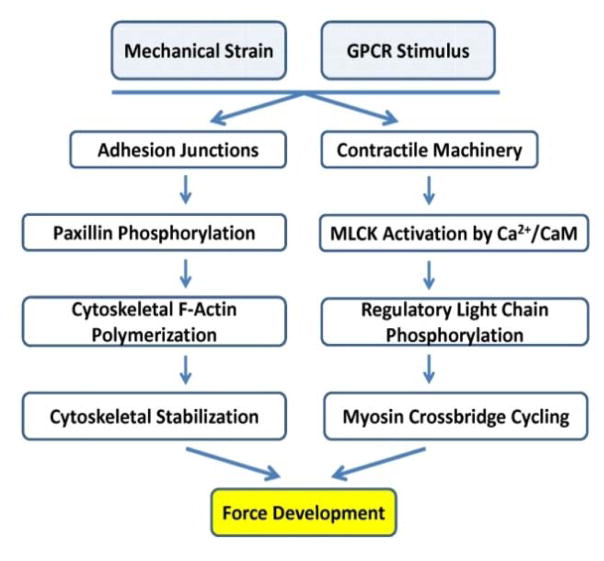
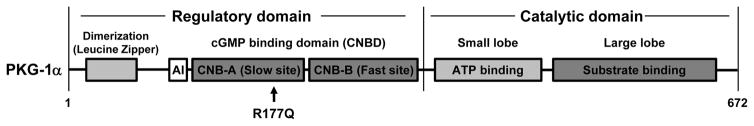
The importance of maintaining contractile function in aortic smooth muscle cells (SMCs) is evident by the fact that heterozygous mutations in the major structural proteins or kinases controlling contraction lead to the formation of aneurysms of the ascending thoracic aorta that predispose to life-threatening aortic dissections. Force generation by SMC requires ATP-dependent cyclic interactions between filaments composed of SMC-specific isoforms of α-actin (encoded by ACTA2) and myosin heavy chain (MYH11). ACTA2 and MYH11 mutations are predicted or have been shown to disrupt this cyclic interaction predispose to thoracic aortic disease. Movement of the myosin motor domain is controlled by phosphorylation of the regulatory light chain on the myosin filament, and loss-of-function mutations in the dedicated kinase for this phosphorylation, myosin light chain kinase (MYLK) also predispose to thoracic aortic disease. Finally, a mutation in the cGMP-activated protein kinase (PRKG1) results in constitutive activation of the kinase in the absence of cGMP, thus driving SMC relaxation in part through increased dephosphorylation of the regulatory light chain and predisposes to thoracic aortic disease. Furthermore, SMCs cannot generate force without connections to the extracellular matrix through focal adhesions, and mutations in the major protein in the extracellular matrix, fibrillin-1, linking SMCs to the matrix also cause thoracic aortic disease in individuals with Marfan syndrome. Thus, disruption of the ability of the aortic SMC to generate force through the elastin-contractile units in response to pulsatile blood flow may be a primary driver for thoracic aortic aneurysms and dissections.
Keywords: Marfan syndrome; aortic aneurysm; aortic dissection; mutation; thoracic aorta; vascular smooth muscle cells.
© 2016 American Heart Association, Inc.
Figures




References
-
- LeMaire SA, Russell L. Epidemiology of thoracic aortic dissection. Nat Rev Cardiol. 2011 Feb;8:103–13. - PubMed
-
- Guo DC, Pannu H, Papke CL, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007 Dec;39:1488–93. - PubMed
-
- Zhu L, Vranckx R, Khau Van KP, Lalande A, Boisset N, Mathieu F, Wegman M, Glancy L, Gasc JM, Brunotte F, Bruneval P, Wolf JE, Michel JB, Jeunemaitre X. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006 Mar;38:343–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
