

AMP-activated Kinase (AMPK) Promotes Innate Immunity and Antiviral Defense through Modulation of Stimulator of Interferon Genes (STING) Signaling
- PMID: 27879319
- PMCID: PMC5217687
- DOI: 10.1074/jbc.M116.763268
AMP-activated Kinase (AMPK) Promotes Innate Immunity and Antiviral Defense through Modulation of Stimulator of Interferon Genes (STING) Signaling
Abstract
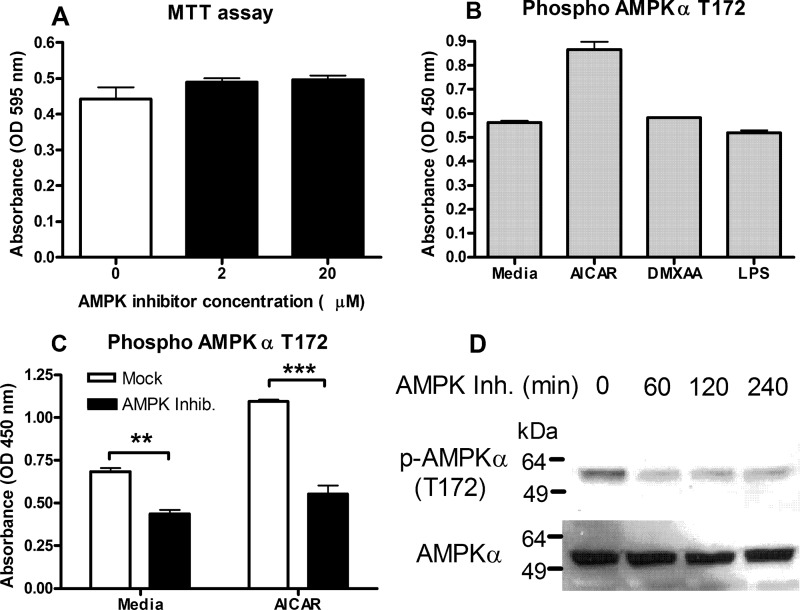
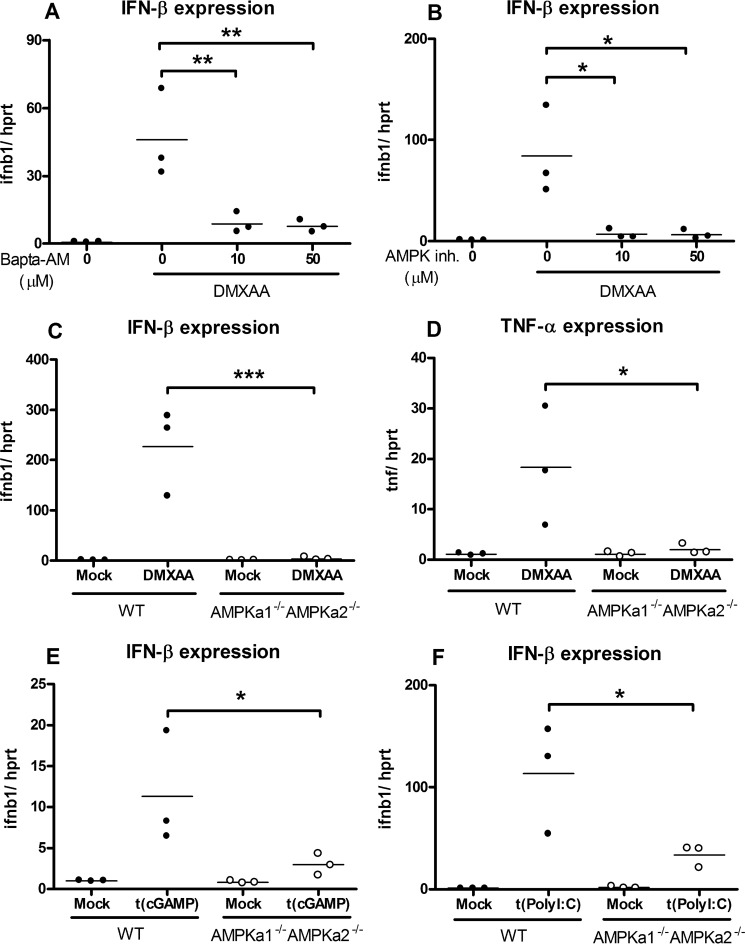
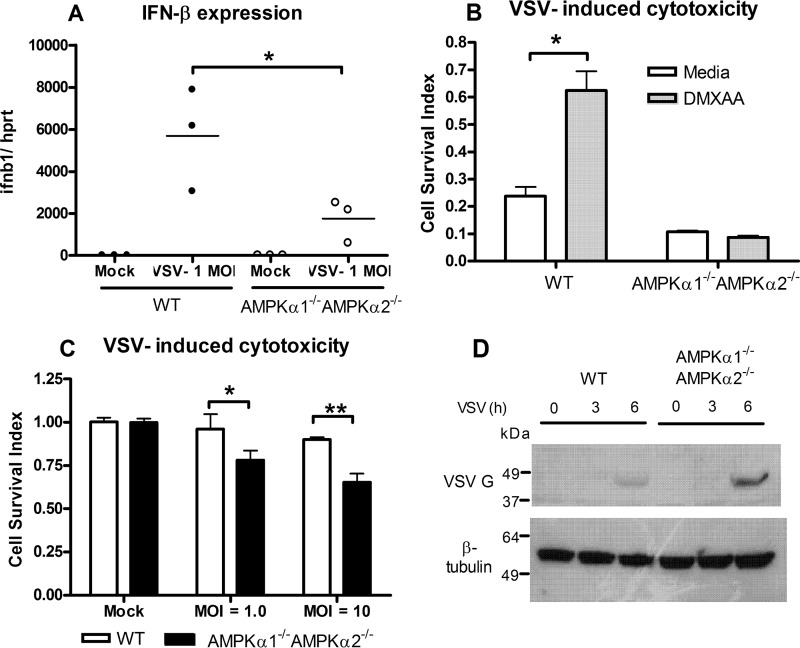
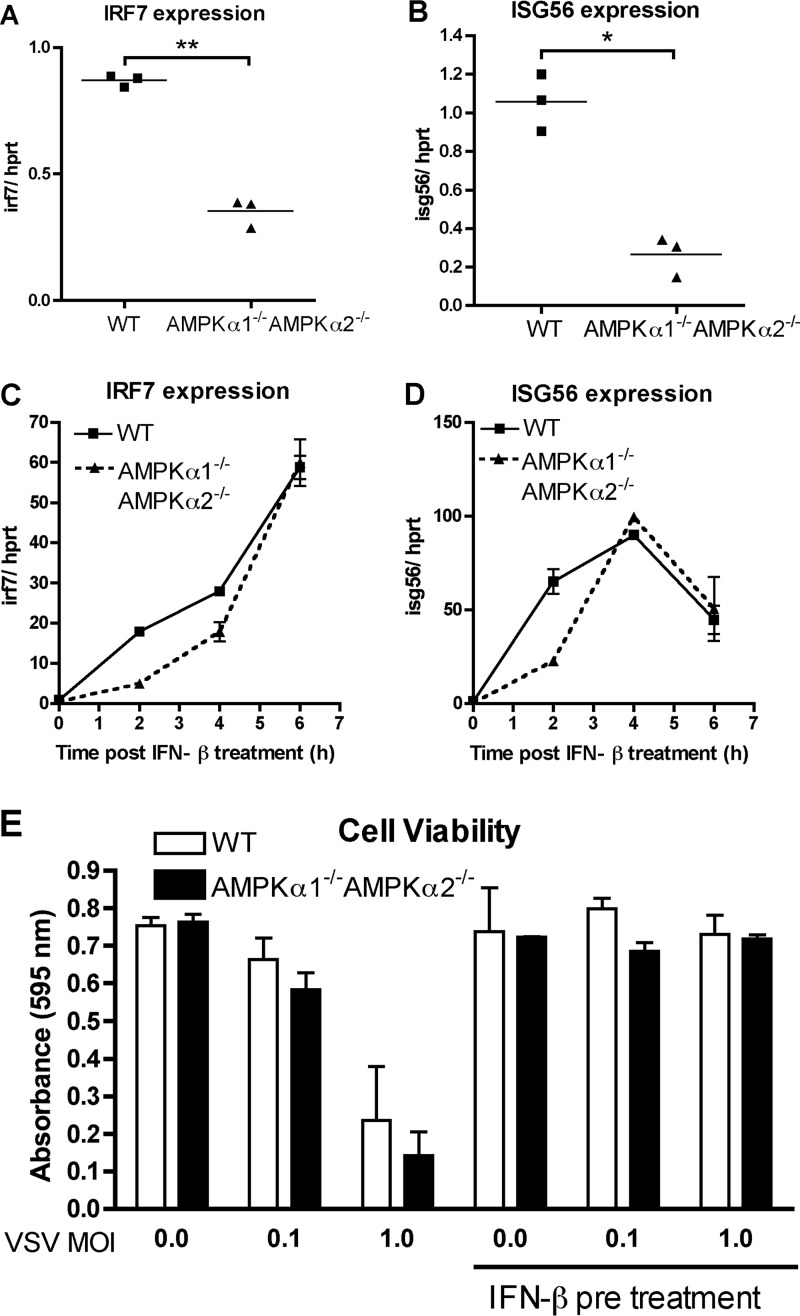
The host protein Stimulator of Interferon Genes (STING) has been shown to be essential for recognition of both viral and intracellular bacterial pathogens, but its regulation remains unclear. Previously, we reported that mitochondrial membrane potential regulates STING-dependent IFN-β induction independently of ATP synthesis. Because mitochondrial membrane potential controls calcium homeostasis, and AMP-activated protein kinase (AMPK) is regulated, in part, by intracellular calcium, we postulated that AMPK participates in STING activation; however, its role has yet to be been defined. Addition of an intracellular calcium chelator or an AMPK inhibitor to either mouse macrophages or mouse embryonic fibroblasts (MEFs) suppressed IFN-β and TNF-α induction following stimulation with the STING-dependent ligand 5,6-dimethyl xanthnone-4-acetic acid (DMXAA). These pharmacological findings were corroborated by showing that MEFs lacking AMPK activity also failed to up-regulate IFN-β and TNF-α after treatment with DMXAA or the natural STING ligand cyclic GMP-AMP (cGAMP). As a result, AMPK-deficient MEFs exhibit impaired control of vesicular stomatitis virus (VSV), a virus sensed by STING that can cause an influenza-like illness in humans. This impairment could be overcome by pretreatment of AMPK-deficient MEFs with type I IFN, illustrating that de novo production of IFN-β in response to VSV plays a key role in antiviral defense during infection. Loss of AMPK also led to dephosphorylation at Ser-555 of the known STING regulator, UNC-51-like kinase 1 (ULK1). However, ULK1-deficient cells responded normally to DMXAA, indicating that AMPK promotes STING-dependent signaling independent of ULK1 in mouse cells.
Keywords: AMP-activated kinase (AMPK); cytokine induction; interferon; macrophage; signal transduction.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures







Similar articles
-
5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator of interferon gene (STING)-dependent innate immune pathways and is regulated by mitochondrial membrane potential.J Biol Chem. 2012 Nov 16;287(47):39776-88. doi: 10.1074/jbc.M112.382986. Epub 2012 Oct 1. J Biol Chem. 2012. PMID: 23027866 Free PMC article.
-
GNAQ Negatively Regulates Antiviral Innate Immune Responses in a Calcineurin-Dependent Manner.J Immunol. 2019 Sep 1;203(5):1288-1297. doi: 10.4049/jimmunol.1900427. Epub 2019 Jul 19. J Immunol. 2019. PMID: 31324725
-
Transfer of cGAMP into Bystander Cells via LRRC8 Volume-Regulated Anion Channels Augments STING-Mediated Interferon Responses and Anti-viral Immunity.Immunity. 2020 May 19;52(5):767-781.e6. doi: 10.1016/j.immuni.2020.03.016. Epub 2020 Apr 10. Immunity. 2020. PMID: 32277911
-
Mechanisms and pathways of innate immune activation and regulation in health and cancer.Hum Vaccin Immunother. 2014;10(11):3270-85. doi: 10.4161/21645515.2014.979640. Hum Vaccin Immunother. 2014. PMID: 25625930 Free PMC article. Review.
-
The cGAS-STING signaling in cardiovascular and metabolic diseases: Future novel target option for pharmacotherapy.Acta Pharm Sin B. 2022 Jan;12(1):50-75. doi: 10.1016/j.apsb.2021.05.011. Epub 2021 May 20. Acta Pharm Sin B. 2022. PMID: 35127372 Free PMC article. Review.
Cited by
-
Innate gene signature distinguishes humoral versus cytotoxic responses to influenza vaccination.J Clin Invest. 2019 Mar 7;129(5):1960-1971. doi: 10.1172/JCI125372. Print 2019 May 1. J Clin Invest. 2019. PMID: 30843873 Free PMC article. Clinical Trial.
-
Cyclic GMP-AMP Synthase (cGAS) Deletion Reduces Severity in Bilateral Nephrectomy Mice through Changes in Neutrophil Extracellular Traps and Mitochondrial Respiration.Biomedicines. 2023 Apr 18;11(4):1208. doi: 10.3390/biomedicines11041208. Biomedicines. 2023. PMID: 37189826 Free PMC article.
-
The interferon-inducible protein TDRD7 inhibits AMP-activated protein kinase and thereby restricts autophagy-independent virus replication.J Biol Chem. 2020 May 15;295(20):6811-6822. doi: 10.1074/jbc.RA120.013533. Epub 2020 Apr 9. J Biol Chem. 2020. PMID: 32273341 Free PMC article.
-
An integrated proteomics approach identifies phosphorylation sites on viral and host proteins that regulate West Nile virus infection.Cell Rep. 2025 May 27;44(5):115728. doi: 10.1016/j.celrep.2025.115728. Epub 2025 May 15. Cell Rep. 2025. PMID: 40381193 Free PMC article.
-
The Antimalaria Drug Artesunate Inhibits Porcine Reproductive and Respiratory Syndrome Virus Replication by Activating AMPK and Nrf2/HO-1 Signaling Pathways.J Virol. 2022 Feb 9;96(3):e0148721. doi: 10.1128/JVI.01487-21. Epub 2021 Nov 17. J Virol. 2022. PMID: 34787456 Free PMC article.
References
-
- Zhong B., Yang Y., Li S., Wang Y. Y., Li Y., Diao F., Lei C., He X., Zhang L., Tien P., and Shu H. B. (2008) The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29, 538–550 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
