

Distribution of Misfolded Prion Protein Seeding Activity Alone Does Not Predict Regions of Neurodegeneration
- PMID: 27880767
- PMCID: PMC5120774
- DOI: 10.1371/journal.pbio.1002579
Distribution of Misfolded Prion Protein Seeding Activity Alone Does Not Predict Regions of Neurodegeneration
Abstract
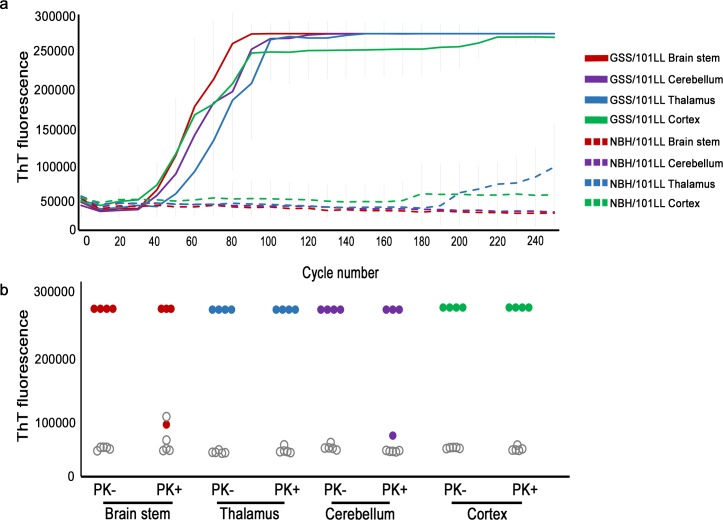
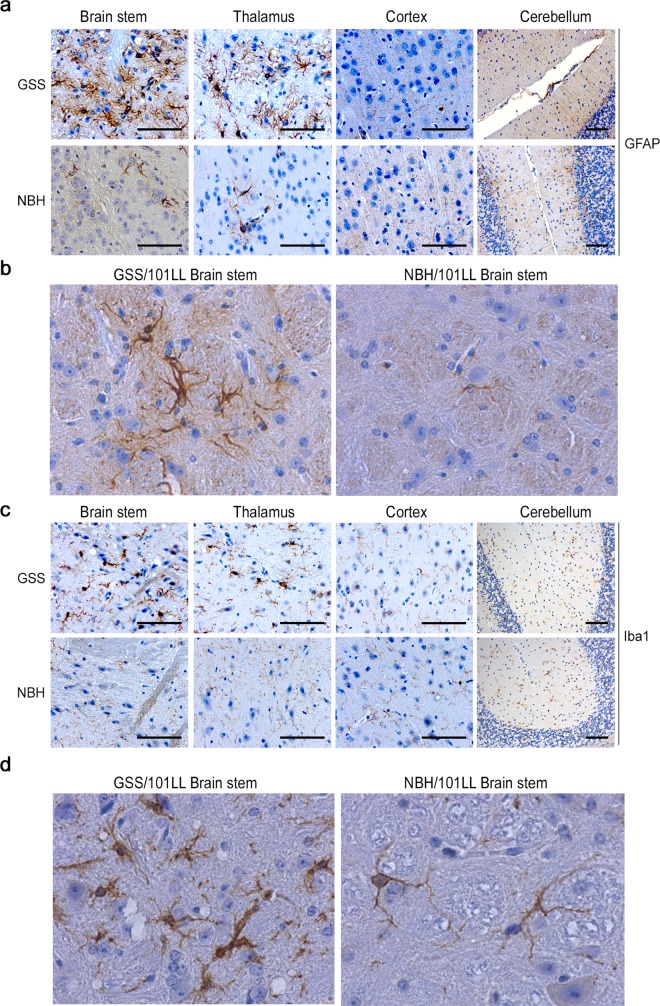
Protein misfolding is common across many neurodegenerative diseases, with misfolded proteins acting as seeds for "prion-like" conversion of normally folded protein to abnormal conformations. A central hypothesis is that misfolded protein accumulation, spread, and distribution are restricted to specific neuronal populations of the central nervous system and thus predict regions of neurodegeneration. We examined this hypothesis using a highly sensitive assay system for detection of misfolded protein seeds in a murine model of prion disease. Misfolded prion protein (PrP) seeds were observed widespread throughout the brain, accumulating in all brain regions examined irrespective of neurodegeneration. Importantly, neither time of exposure nor amount of misfolded protein seeds present determined regions of neurodegeneration. We further demonstrate two distinct microglia responses in prion-infected brains: a novel homeostatic response in all regions and an innate immune response restricted to sites of neurodegeneration. Therefore, accumulation of misfolded prion protein alone does not define targeting of neurodegeneration, which instead results only when misfolded prion protein accompanies a specific innate immune response.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures






References
-
- Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, et al. (1993) Mice devoid of PrP are resistant to scrapie. Cell 73: 1339–1347. - PubMed
-
- Cunningham C, Deacon R, Wells H, Boche D, Waters S, et al. (2003) Synaptic changes characterize early behavioural signs in the ME7 model of murine prion disease. Eur J Neurosci 17: 2147–2155. - PubMed
MeSH terms
Substances
Grants and funding
- BBS/E/D/20251967/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BBS/E/D/20251968/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BBS/E/D/20211551/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BBS/E/D/20211552/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/F003722/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
