

Activation of COX-2/PGE2 Promotes Sapovirus Replication via the Inhibition of Nitric Oxide Production
- PMID: 27881647
- PMCID: PMC5244346
- DOI: 10.1128/JVI.01656-16
Activation of COX-2/PGE2 Promotes Sapovirus Replication via the Inhibition of Nitric Oxide Production
Abstract
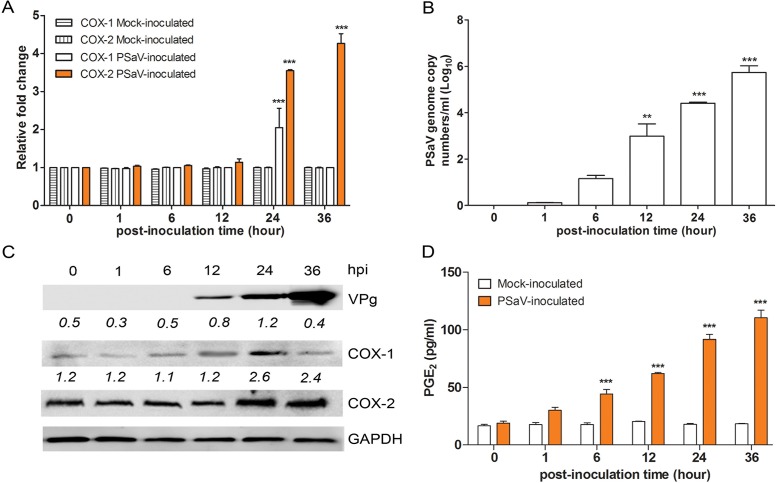
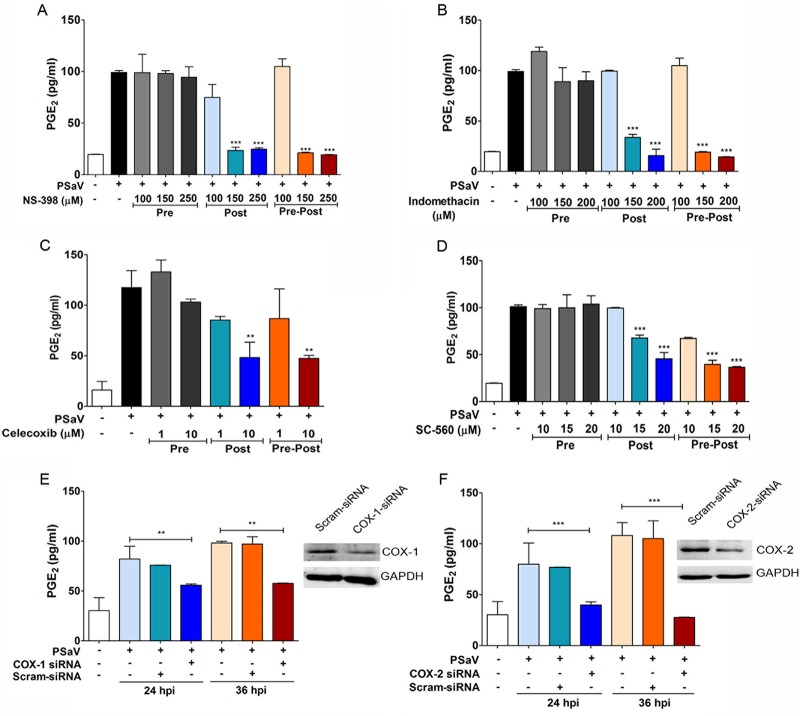
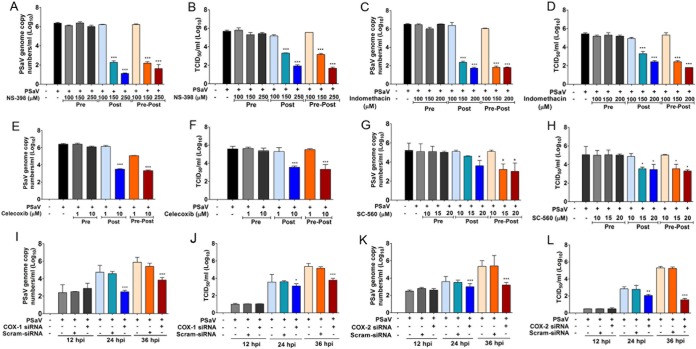
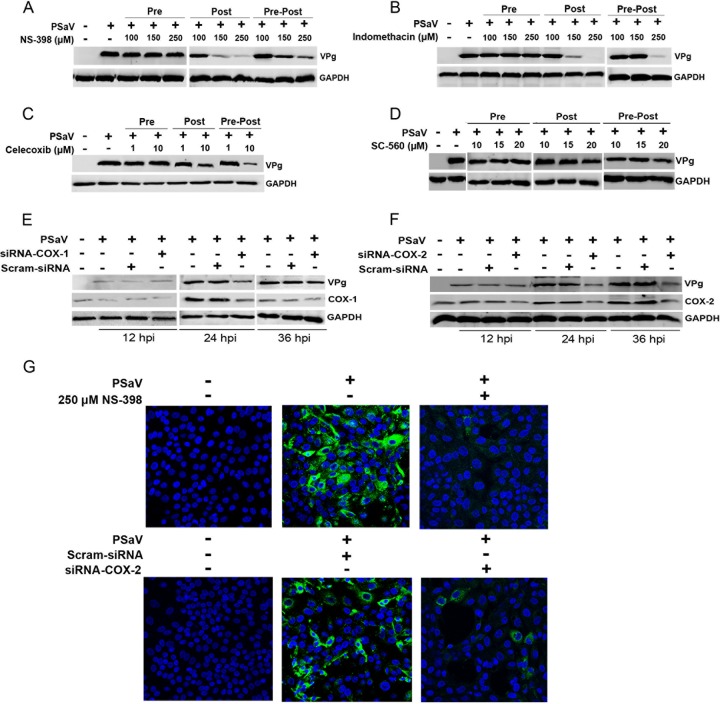
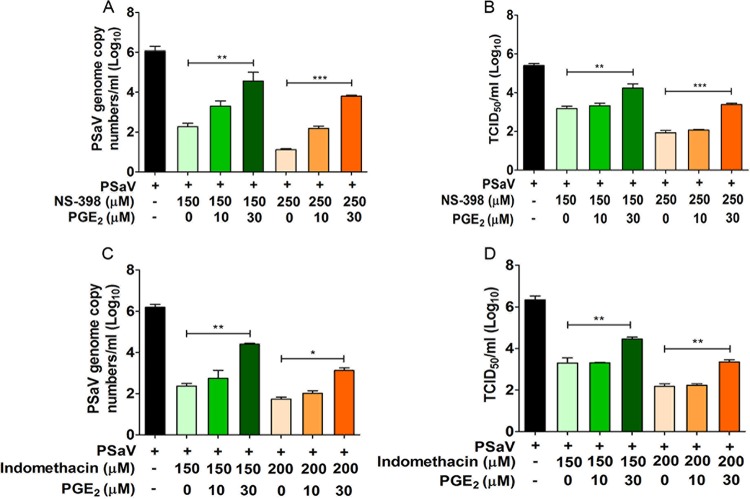
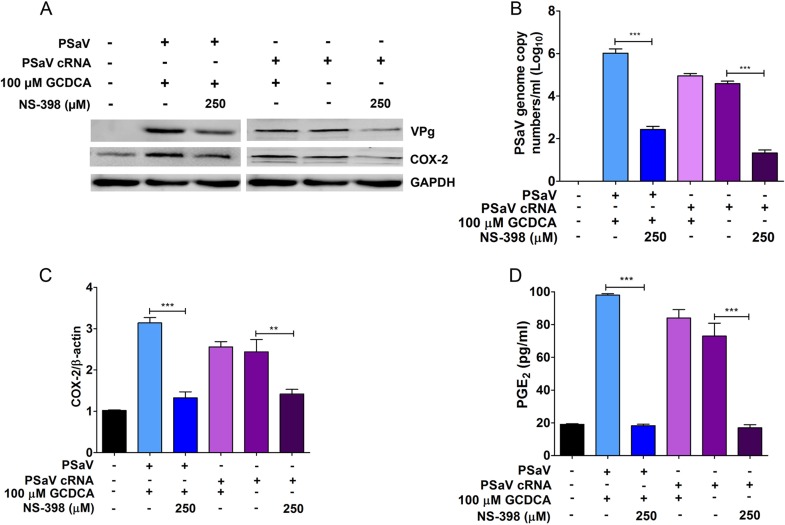
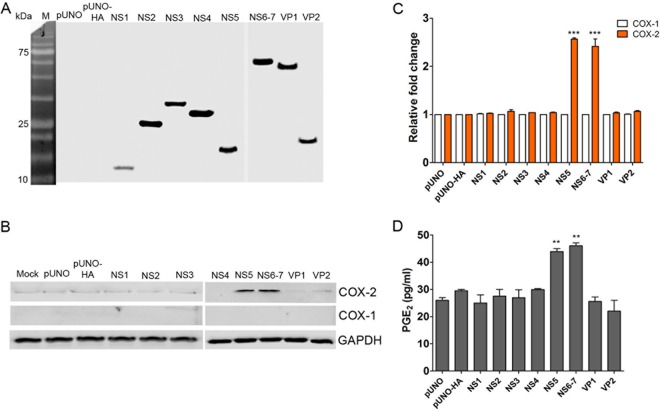
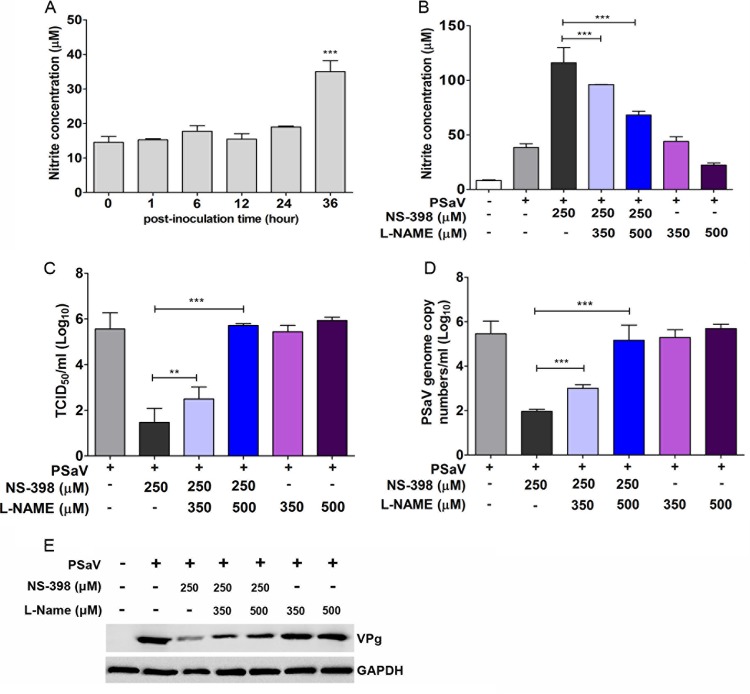
Enteric caliciviruses in the genera Norovirus and Sapovirus are important pathogens that cause severe acute gastroenteritis in both humans and animals. Cyclooxygenases (COXs) and their final product, prostaglandin E2 (PGE2), are known to play important roles in the modulation of both the host response to infection and the replicative cycles of several viruses. However, the precise mechanism(s) by which the COX/PGE2 pathway regulates sapovirus replication remains largely unknown. In this study, infection with porcine sapovirus (PSaV) strain Cowden, the only cultivable virus within the genus Sapovirus, markedly increased COX-2 mRNA and protein levels at 24 and 36 h postinfection (hpi), with only a transient increase in COX-1 levels seen at 24 hpi. The treatment of cells with pharmacological inhibitors, such as nonsteroidal anti-inflammatory drugs or small interfering RNAs (siRNAs) against COX-1 and COX-2, significantly reduced PGE2 production, as well as PSaV replication. Expression of the viral proteins VPg and ProPol was associated with activation of the COX/PGE2 pathway. We observed that pharmacological inhibition of COX-2 dramatically increased NO production, causing a reduction in PSaV replication that could be restored by inhibition of nitric oxide synthase via the inhibitor N-nitro-l-methyl-arginine ester. This study identified a pivotal role for the COX/PGE2 pathway in the regulation of NO production during the sapovirus life cycle, providing new insights into the life cycle of this poorly characterized family of viruses. Our findings also reveal potential new targets for treatment of sapovirus infection.
Importance: Sapoviruses are among the major etiological agents of acute gastroenteritis in both humans and animals, but little is known about sapovirus host factor requirements. Here, using only cultivable porcine sapovirus (PSaV) strain Cowden, we demonstrate that PSaV induced the vitalization of the cyclooxygenase (COX) and prostaglandin E2 (PGE2) pathway. Targeting of COX-1/2 using nonsteroidal anti-inflammatory drugs (NSAIDs) such as the COX-1/2 inhibitor indomethacin and the COX-2-specific inhibitors NS-398 and celecoxib or siRNAs targeting COXs, inhibited PSaV replication. Expression of the viral proteins VPg and ProPol was associated with activation of the COX/PGE2 pathway. We further demonstrate that the production of PGE2 provides a protective effect against the antiviral effector mechanism of nitric oxide. Our findings uncover a new mechanism by which PSaV manipulates the host cell to provide an environment suitable for efficient viral growth, which in turn can be a new target for treatment of sapovirus infection.
Keywords: caliciviruses; cyclooxygenases; nitric oxide; prostaglandin E2; sapovirus.
Copyright © 2017 Alfajaro et al.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
