

Lipid disequilibrium disrupts ER proteostasis by impairing ERAD substrate glycan trimming and dislocation
- PMID: 27881664
- PMCID: PMC5231896
- DOI: 10.1091/mbc.E16-07-0483
Lipid disequilibrium disrupts ER proteostasis by impairing ERAD substrate glycan trimming and dislocation
Abstract
The endoplasmic reticulum (ER) mediates the folding, maturation, and deployment of the secretory proteome. Proteins that fail to achieve their native conformation are retained in the ER and targeted for clearance by ER-associated degradation (ERAD), a sophisticated process that mediates the ubiquitin-dependent delivery of substrates to the 26S proteasome for proteolysis. Recent findings indicate that inhibition of long-chain acyl-CoA synthetases with triacsin C, a fatty acid analogue, impairs lipid droplet (LD) biogenesis and ERAD, suggesting a role for LDs in ERAD. However, whether LDs are involved in the ERAD process remains an outstanding question. Using chemical and genetic approaches to disrupt diacylglycerol acyltransferase (DGAT)-dependent LD biogenesis, we provide evidence that LDs are dispensable for ERAD in mammalian cells. Instead, our results suggest that triacsin C causes global alterations in the cellular lipid landscape that disrupt ER proteostasis by interfering with the glycan trimming and dislocation steps of ERAD. Prolonged triacsin C treatment activates both the IRE1 and PERK branches of the unfolded protein response and ultimately leads to IRE1-dependent cell death. These findings identify an intimate relationship between fatty acid metabolism and ER proteostasis that influences cell viability.
© 2017 To, Peterson, et al. This article is distributed by The American Society for Cell Biology under license from the author(s). Two months after publication it is available to the public under an Attribution–Noncommercial–Share Alike 3.0 Unported Creative Commons License (http://creativecommons.org/licenses/by-nc-sa/3.0).
Figures
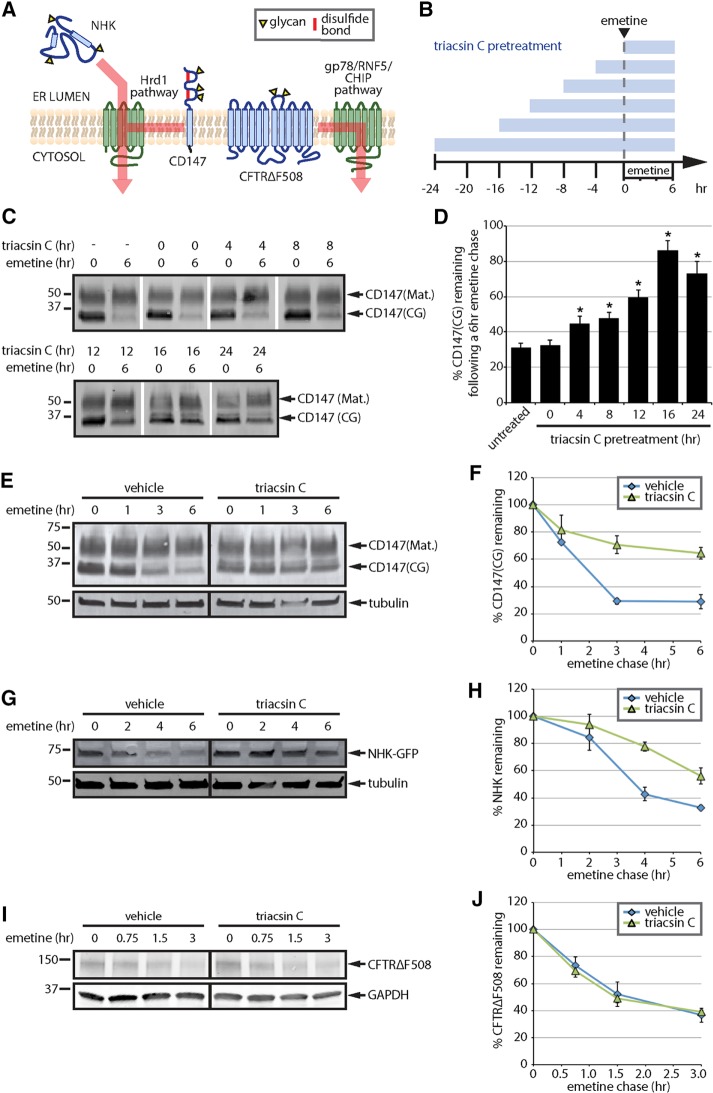
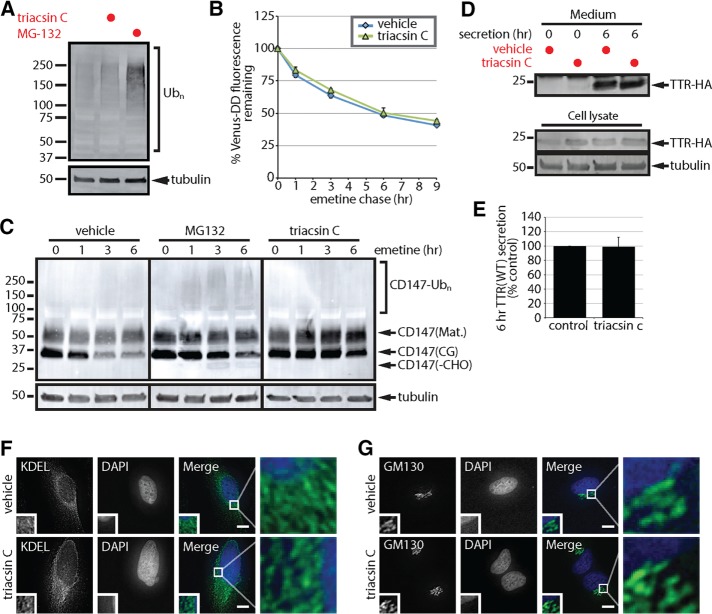
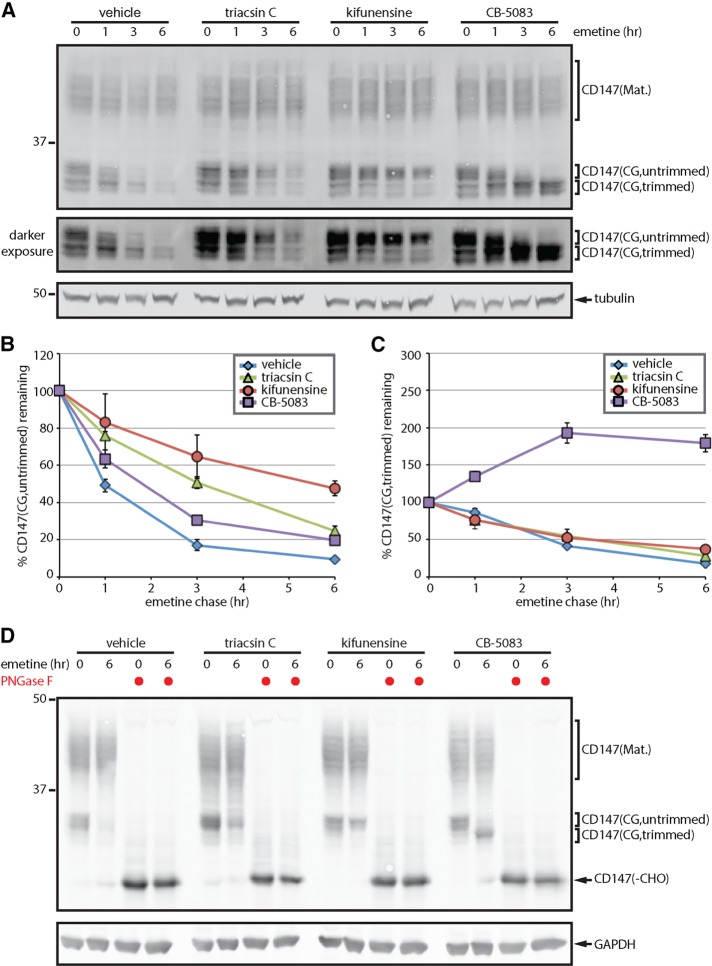
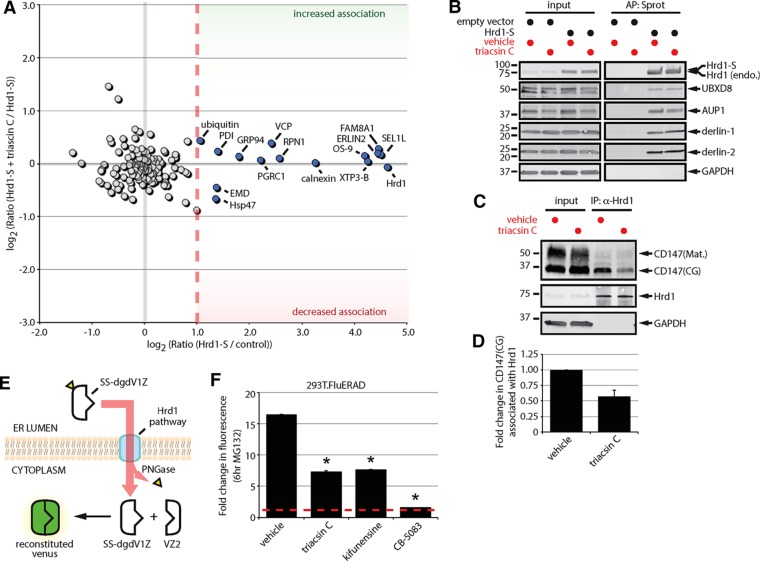
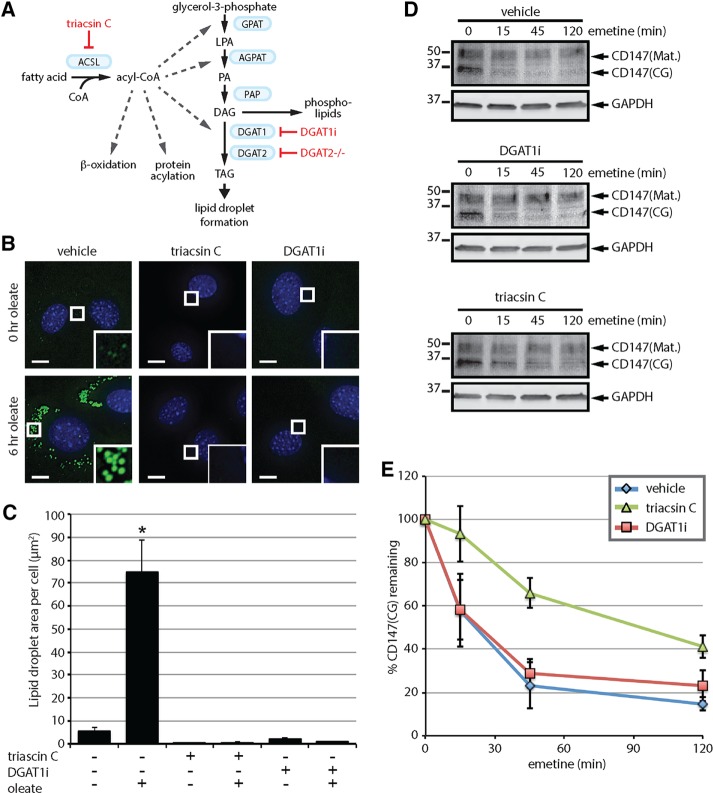
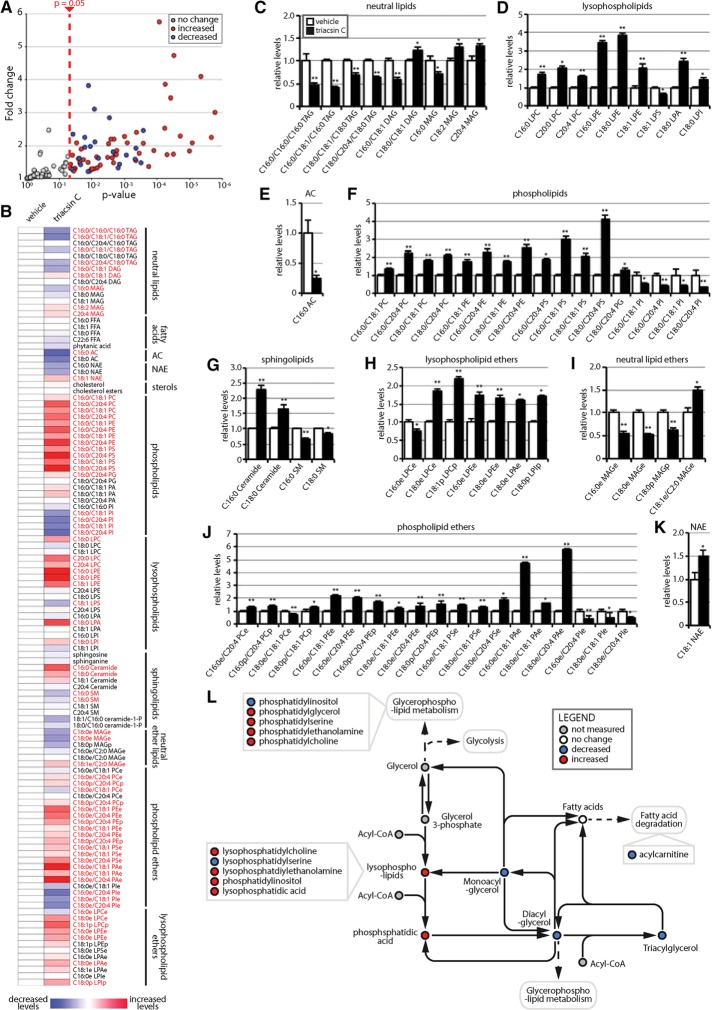
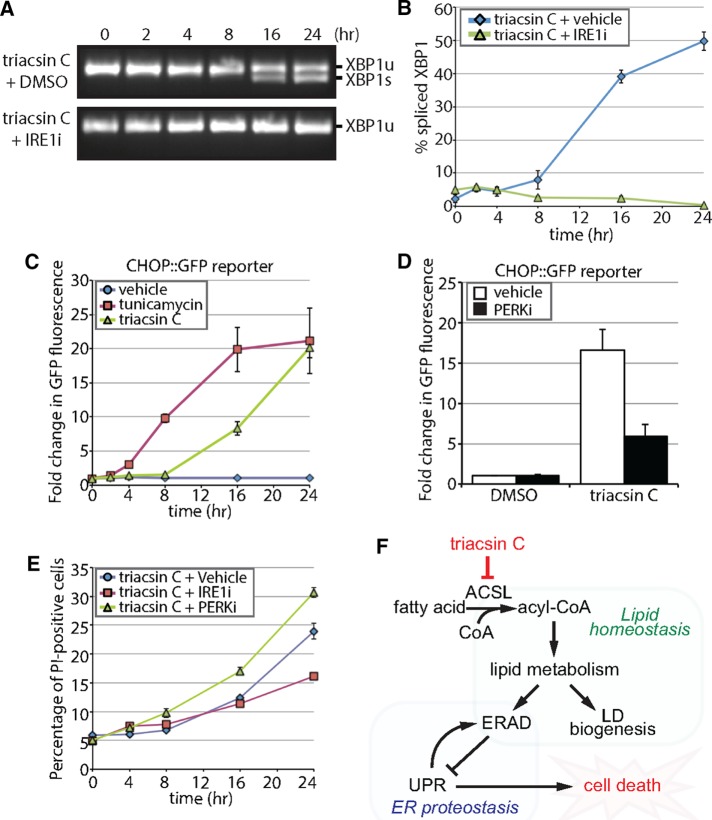
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
