

Localization of 1-deoxysphingolipids to mitochondria induces mitochondrial dysfunction
- PMID: 27881717
- PMCID: PMC5234710
- DOI: 10.1194/jlr.M068676
Localization of 1-deoxysphingolipids to mitochondria induces mitochondrial dysfunction
Abstract
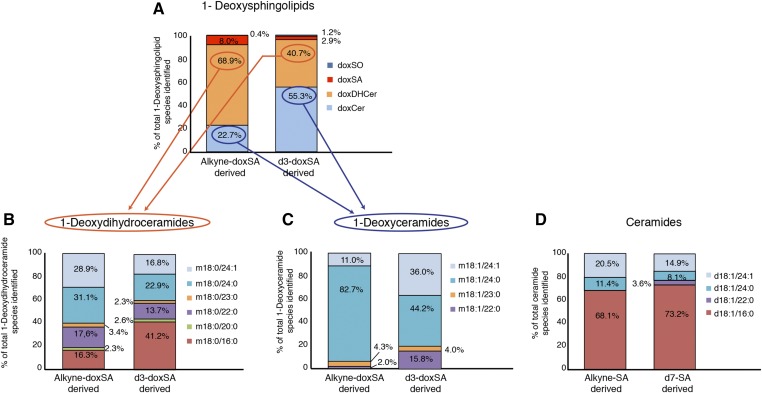
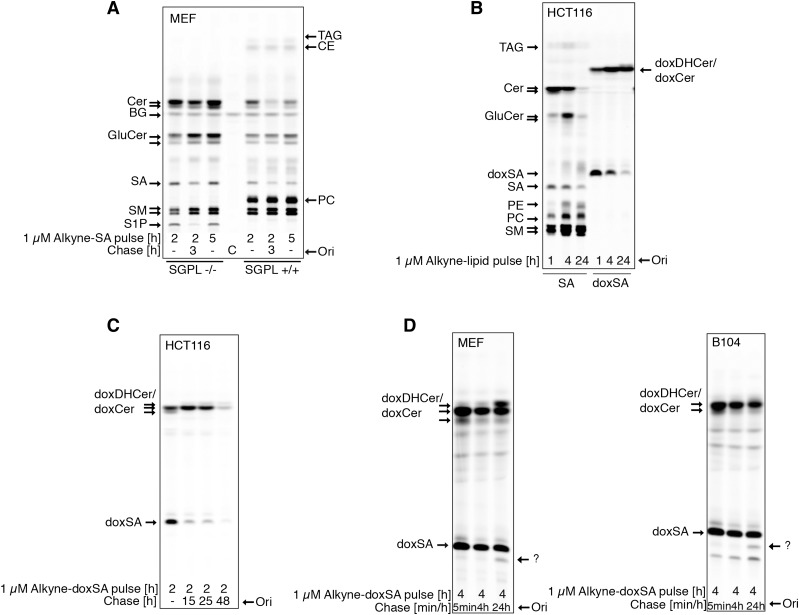
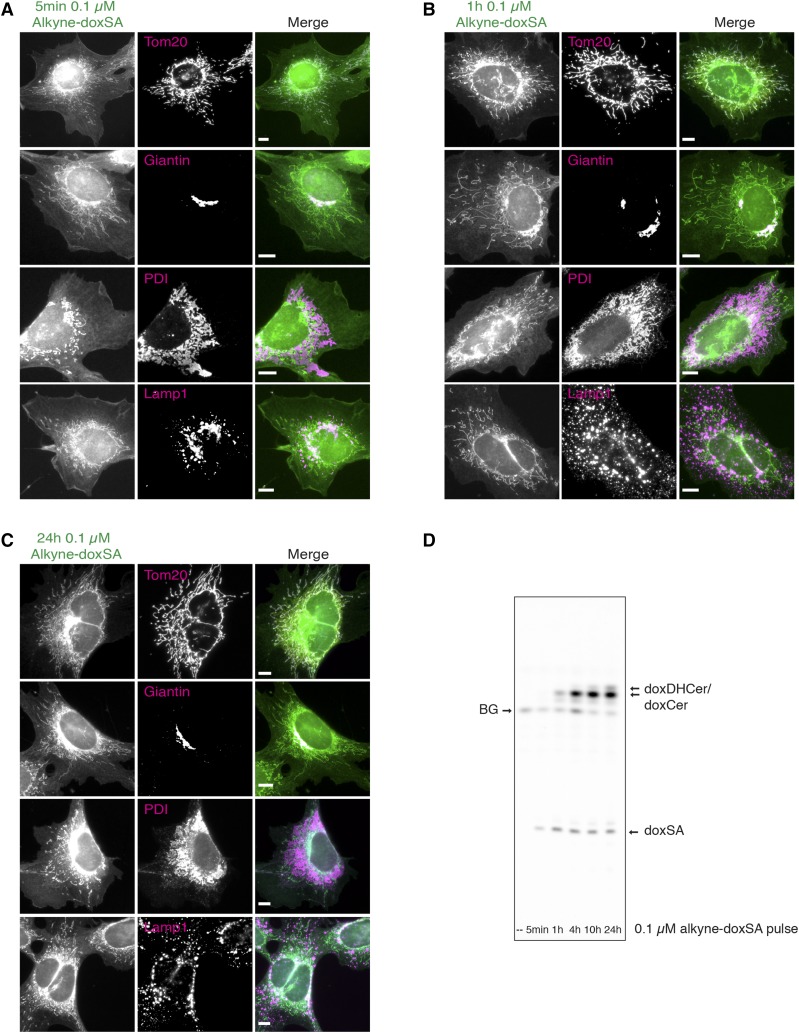
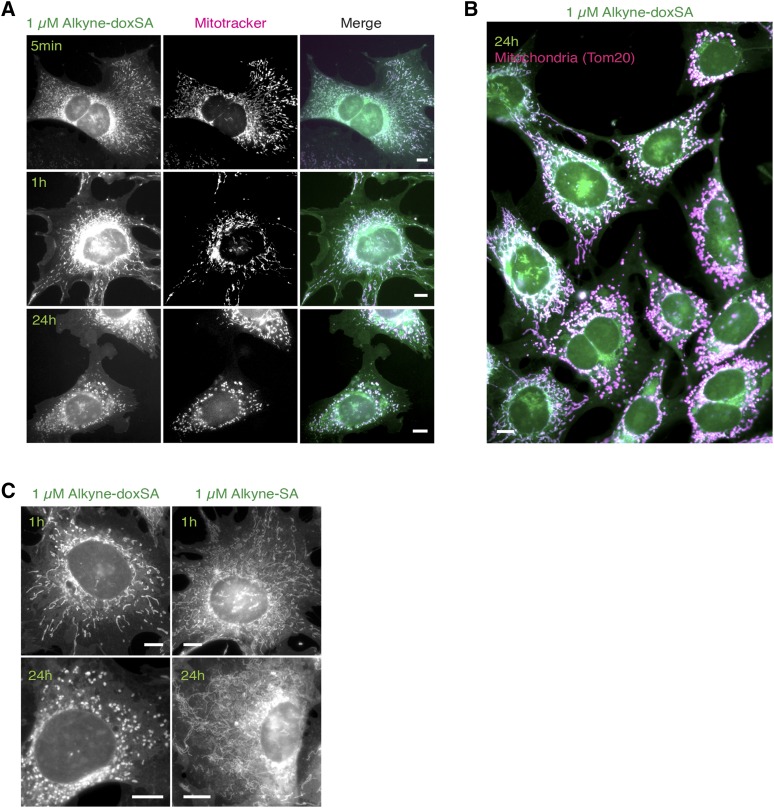
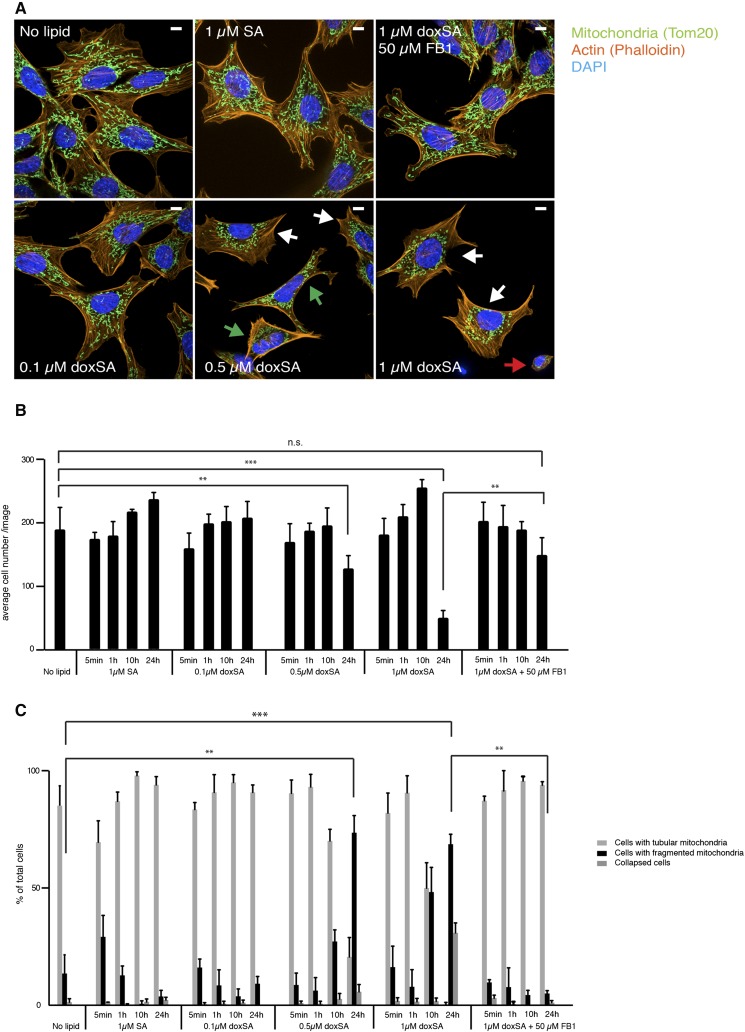
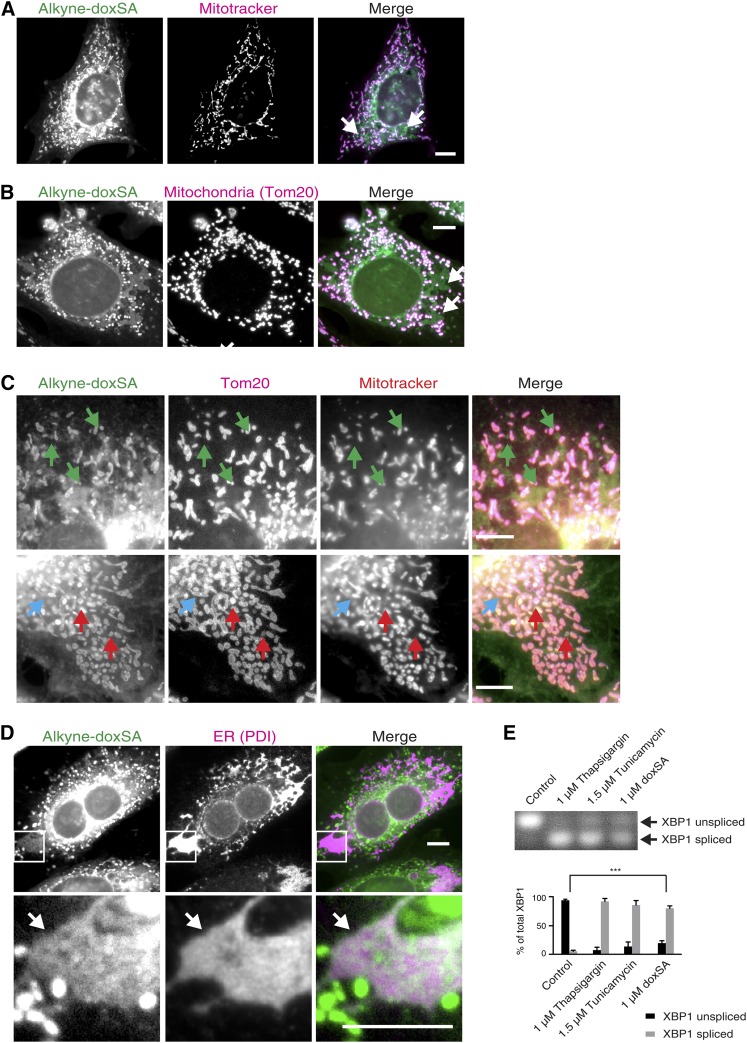
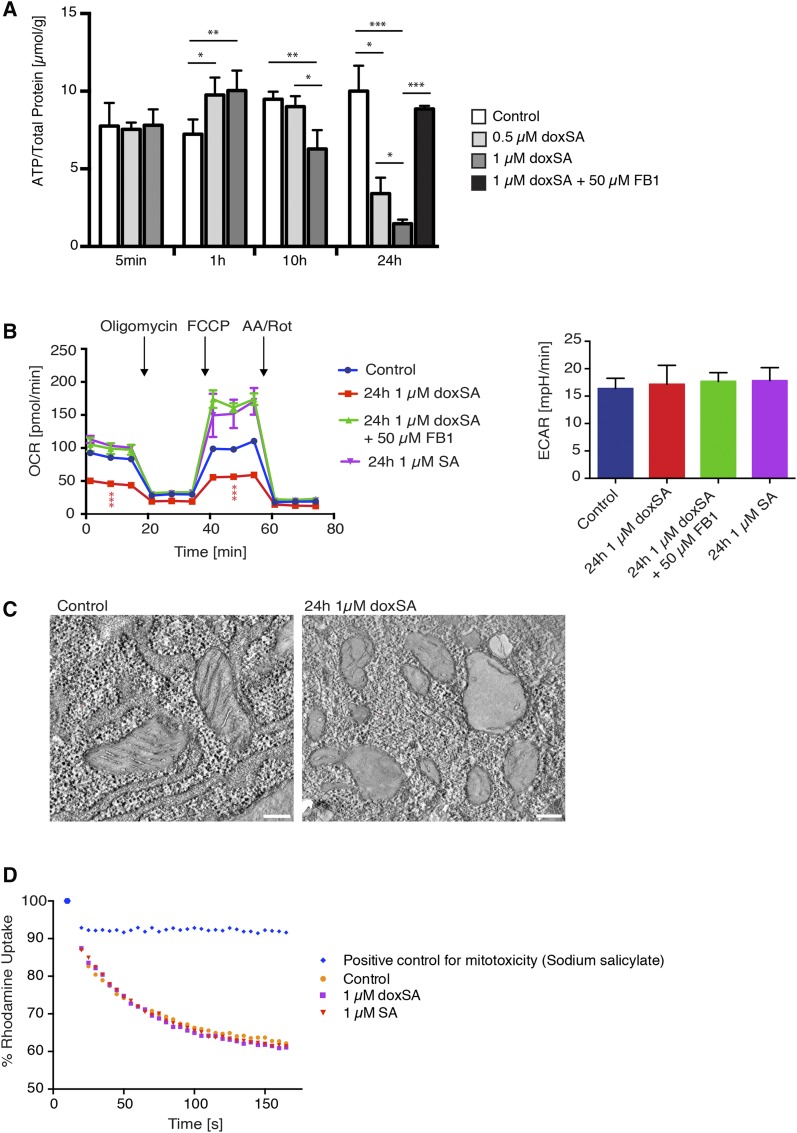
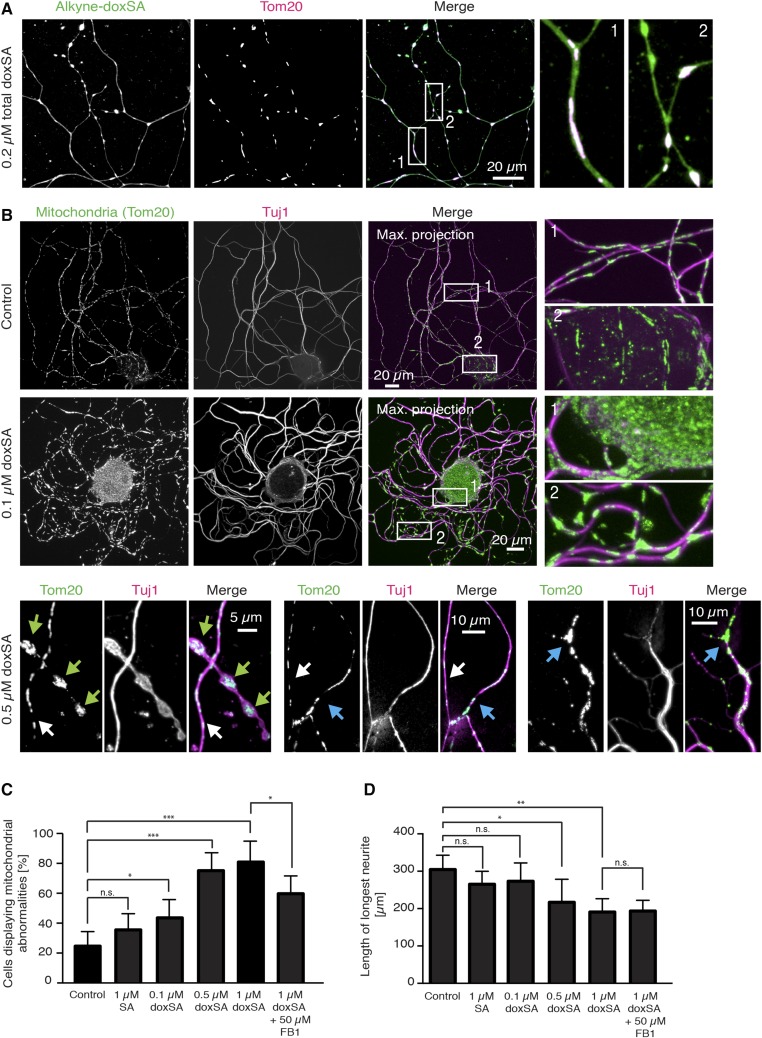
1-Deoxysphingolipids (deoxySLs) are atypical sphingolipids that are elevated in the plasma of patients with type 2 diabetes and hereditary sensory and autonomic neuropathy type 1 (HSAN1). Clinically, diabetic neuropathy and HSAN1 are very similar, suggesting the involvement of deoxySLs in the pathology of both diseases. However, very little is known about the biology of these lipids and the underlying pathomechanism. We synthesized an alkyne analog of 1-deoxysphinganine (doxSA), the metabolic precursor of all deoxySLs, to trace the metabolism and localization of deoxySLs. Our results indicate that the metabolism of these lipids is restricted to only some lipid species and that they are not converted to canonical sphingolipids or fatty acids. Furthermore, exogenously added alkyne-doxSA [(2S,3R)-2-aminooctadec-17-yn-3-ol] localized to mitochondria, causing mitochondrial fragmentation and dysfunction. The induced mitochondrial toxicity was also shown for natural doxSA, but not for sphinganine, and was rescued by inhibition of ceramide synthase activity. Our findings therefore indicate that mitochondrial enrichment of an N-acylated doxSA metabolite may contribute to the neurotoxicity seen in diabetic neuropathy and HSAN1. Hence, we provide a potential explanation for the characteristic vulnerability of peripheral nerves to elevated levels of deoxySLs.
Keywords: ES-285; chemical synthesis; diabetes; inborn errors of metabolism; lipids/chemistry; metabolic syndrome; mitotoxicity; neurons; peripheral neuropathy; sphingolipids.
Copyright © 2017 by the American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Zitomer N. C., Mitchell T., Voss K. A., Bondy G. S., Pruett S. T., Garnier-Amblard E. C., Liebeskind L. S., Park H., Wang E., Sullards M. C., et al. . 2009. Ceramide synthase inhibition by fumonisin B1 causes accumulation of 1-deoxysphinganine: a novel category of bioactive 1-deoxysphingoid bases and 1-deoxydihydroceramides biosynthesized by mammalian cell lines and animals. J. Biol. Chem. 284: 4786–4795. - PMC - PubMed
-
- Houlden H., King R., Blake J., Groves M., Love S., Woodward C., Hammans S., Nicoll J., Lennox G., O’Donovan D. G., et al. . 2006. Clinical, pathological and genetic characterization of hereditary sensory and autonomic neuropathy type 1 (HSAN I). Brain. 129: 411–425. - PubMed
-
- Bejaoui K., Wu C., Scheffler M. D., Haan G., Ashby P., Wu L., de Jong P., and Brown R. H. Jr. 2001. SPTLC1 is mutated in hereditary sensory neuropathy, type 1. Nat. Genet. 27: 261–262. - PubMed
-
- Dawkins J. L., Hulme D. J., Brahmbhatt S. B., Auer-Grumbach M., and Nicholson G. A.. 2001. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat. Genet. 27: 309–312. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
