

Novel Roles for the Insulin-Regulated Glucose Transporter-4 in Hippocampally Dependent Memory
- PMID: 27881773
- PMCID: PMC5125244
- DOI: 10.1523/JNEUROSCI.1700-16.2016
Novel Roles for the Insulin-Regulated Glucose Transporter-4 in Hippocampally Dependent Memory
Abstract
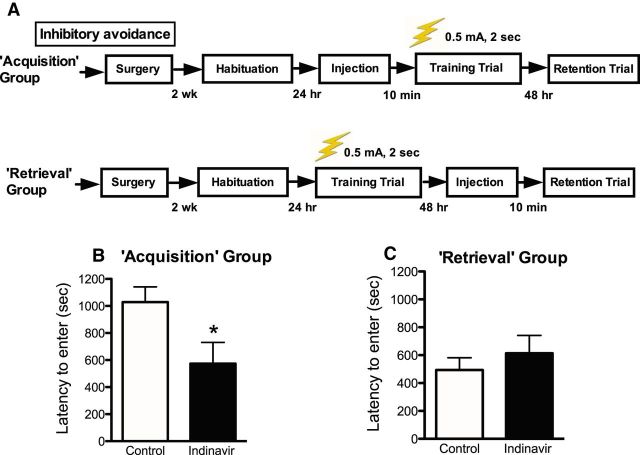
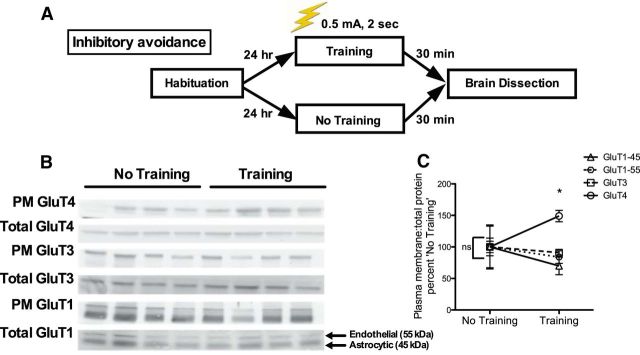
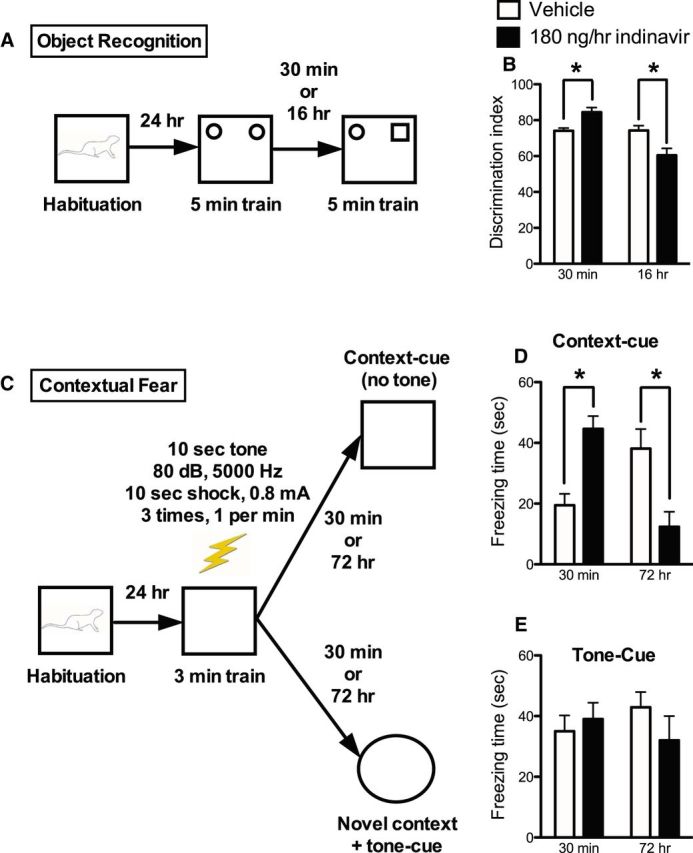
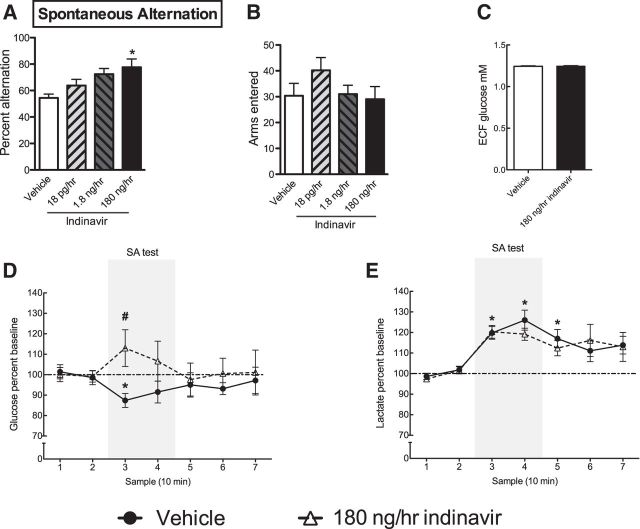
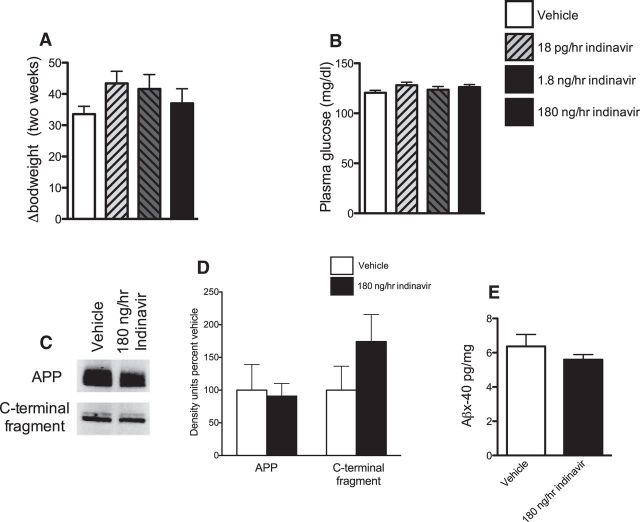
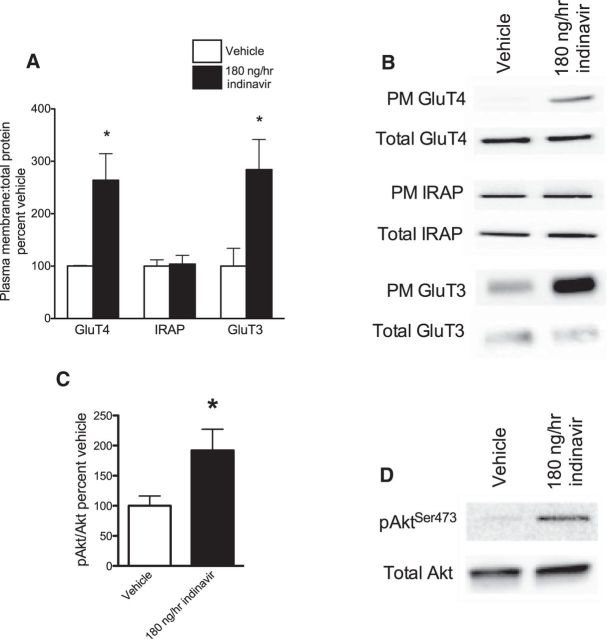
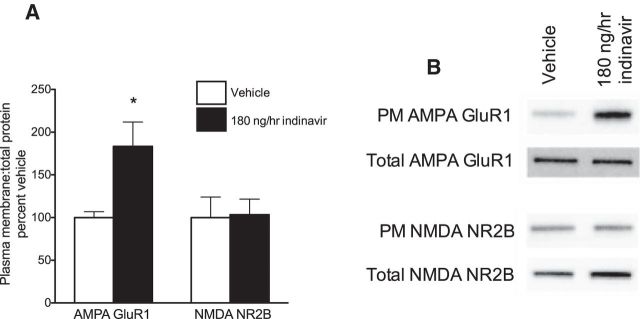
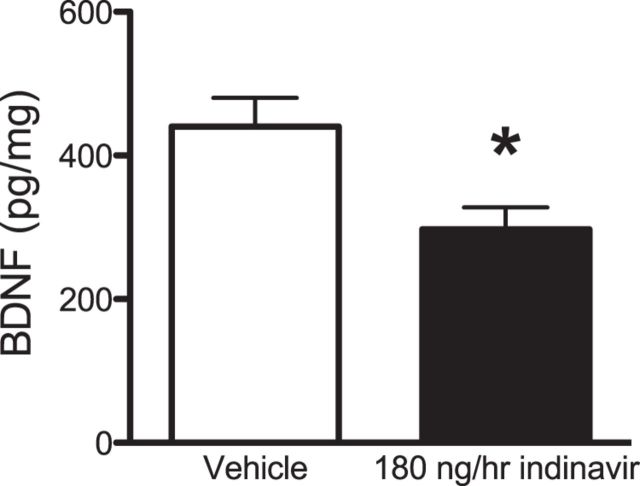
The insulin-regulated glucose transporter-4 (GluT4) is critical for insulin- and contractile-mediated glucose uptake in skeletal muscle. GluT4 is also expressed in some hippocampal neurons, but its functional role in the brain is unclear. Several established molecular modulators of memory processing regulate hippocampal GluT4 trafficking and hippocampal memory formation is limited by both glucose metabolism and insulin signaling. Therefore, we hypothesized that hippocampal GluT4 might be involved in memory processes. Here, we show that, in male rats, hippocampal GluT4 translocates to the plasma membrane after memory training and that acute, selective intrahippocampal inhibition of GluT4-mediated glucose transport impaired memory acquisition, but not memory retrieval. Other studies have shown that prolonged systemic GluT4 blockade causes insulin resistance. Unexpectedly, we found that prolonged hippocampal blockade of glucose transport through GluT4-upregulated markers of hippocampal insulin signaling prevented task-associated depletion of hippocampal glucose and enhanced both working and short-term memory while also impairing long-term memory. These effects were accompanied by increased expression of hippocampal AMPA GluR1 subunits and the neuronal GluT3, but decreased expression of hippocampal brain-derived neurotrophic factor, consistent with impaired ability to form long-term memories. Our findings are the first to show the cognitive impact of brain GluT4 modulation. They identify GluT4 as a key regulator of hippocampal memory processing and also suggest differential regulation of GluT4 in the hippocampus from that in peripheral tissues.
Significance statement: The role of insulin-regulated glucose transporter-4 (GluT4) in the brain is unclear. In the current study, we demonstrate that GluT4 is a critical component of hippocampal memory processes. Memory training increased hippocampal GluT4 translocation and memory acquisition was impaired by GluT4 blockade. Unexpectedly, whereas long-term inhibition of GluT4 impaired long-term memory, short-term memory was enhanced. These data further our understanding of the molecular mechanisms of memory and have particular significance for type 2 diabetes (in which GluT4 activity in the periphery is impaired) and Alzheimer's disease (which is linked to impaired brain insulin signaling and for which type 2 diabetes is a key risk factor). Both diseases cause marked impairment of hippocampal memory linked to hippocampal hypometabolism, suggesting the possibility that brain GluT4 dysregulation may be one cause of cognitive impairment in these disease states.
Keywords: GluT4; glucose; hippocampus; memory.
Copyright © 2016 the authors 0270-6474/16/3611851-14$15.00/0.
Figures
References
-
- Alquier T, Leloup C, Lorsignol A, Pénicaud L. Translocable glucose transporters in the brain. Diabetes. 2006;55:S131–S138. doi: 10.2337/db06-S021. - DOI
-
- Anagnostaras SG, Gale GD, Fanselow MS. Hippocampus and contextual fear conditioning: recent controversies and advances. Hippocampus. 2001;11:8–17. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical