

Two tissue-resident progenitor lineages drive distinct phenotypes of heterotopic ossification
- PMID: 27881824
- PMCID: PMC6407419
- DOI: 10.1126/scitranslmed.aaf1090
Two tissue-resident progenitor lineages drive distinct phenotypes of heterotopic ossification
Abstract
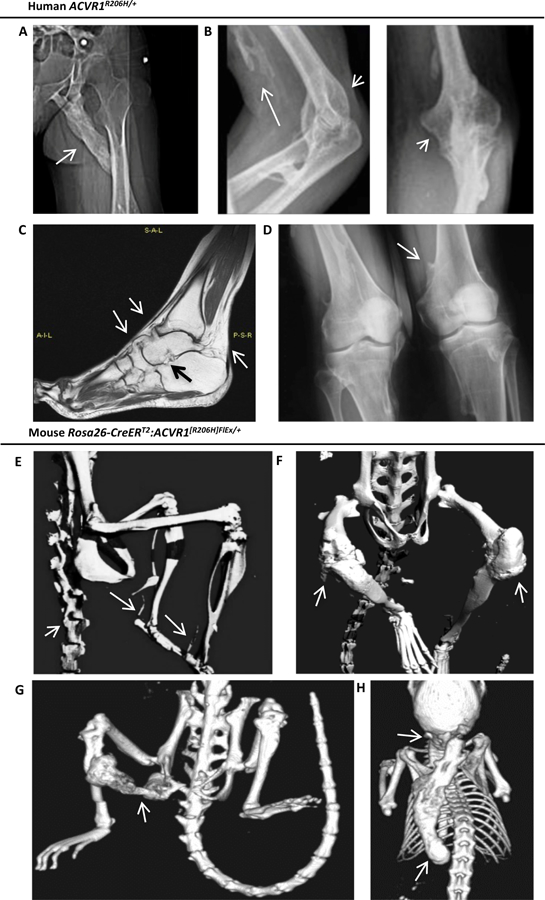
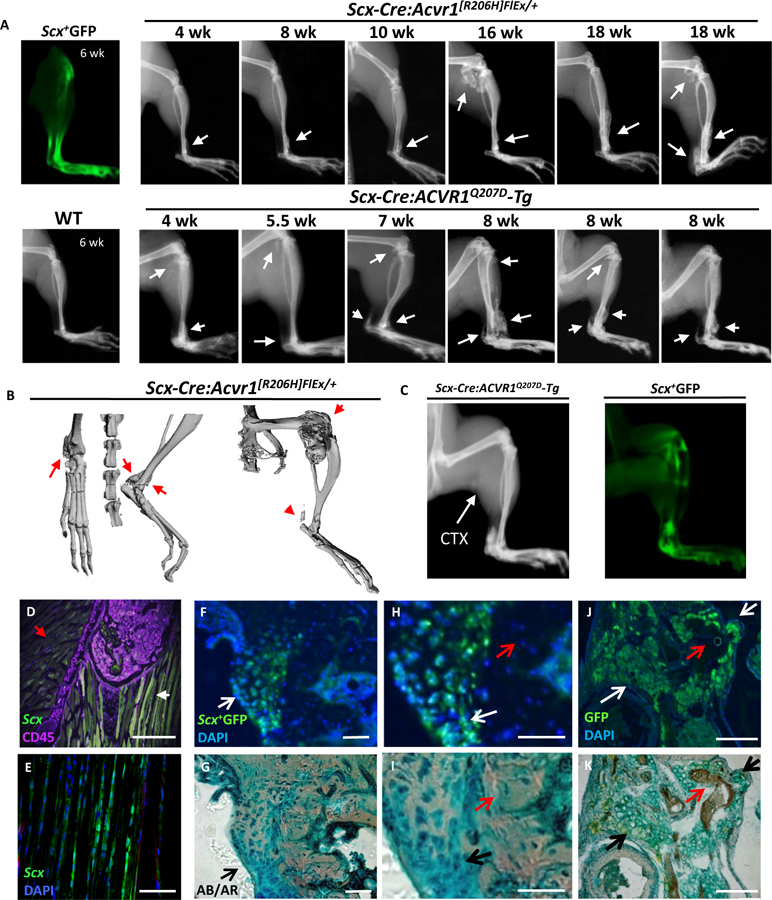
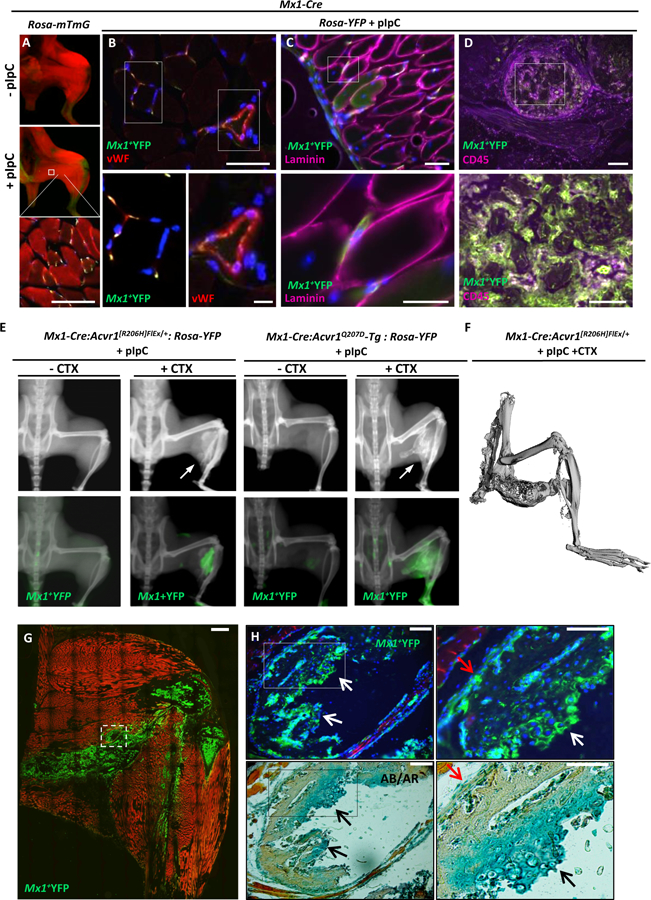
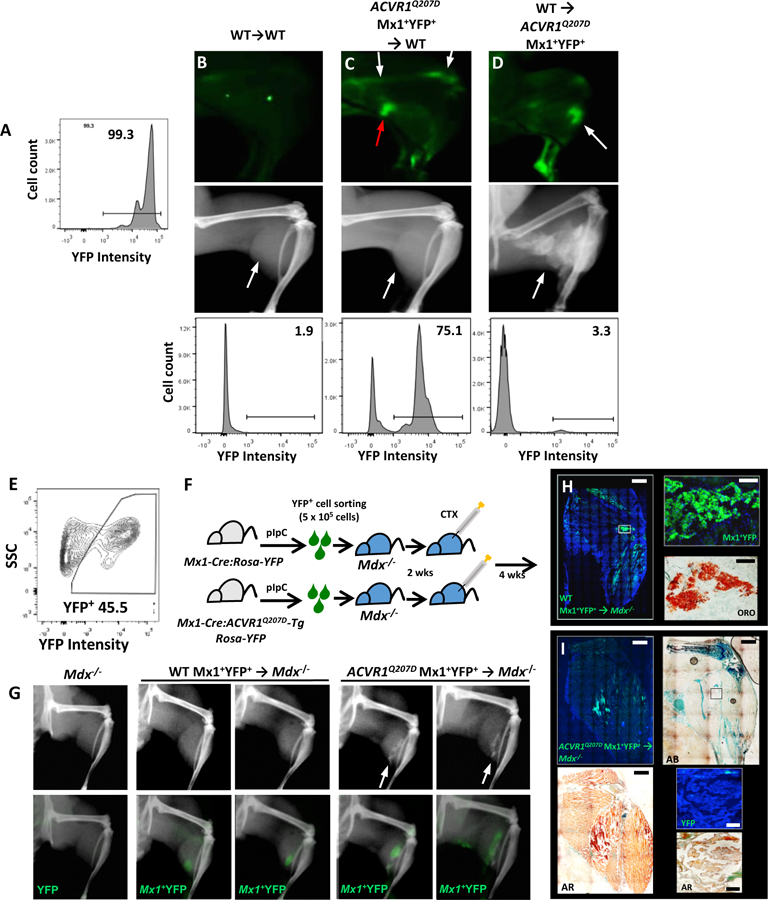
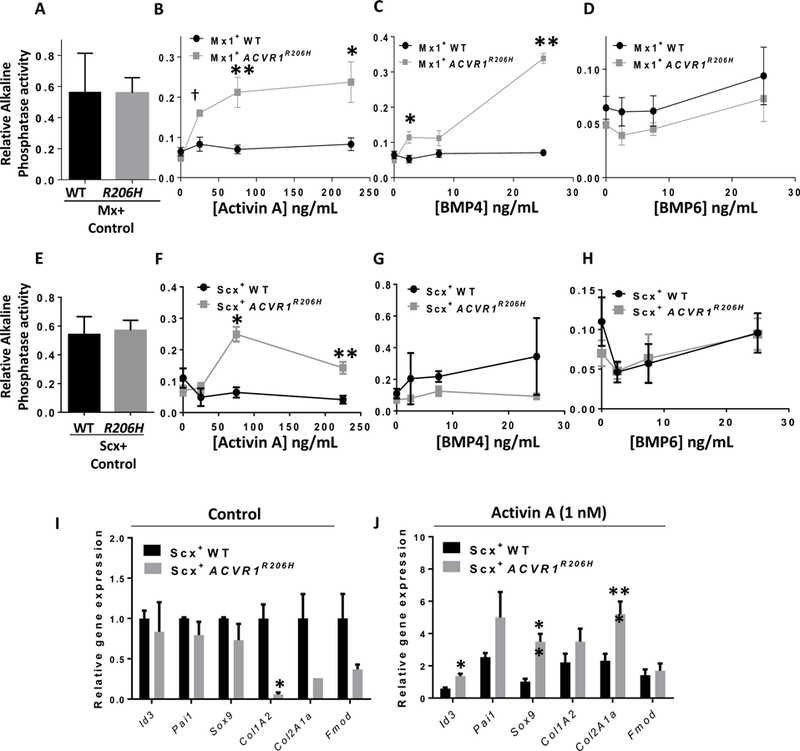
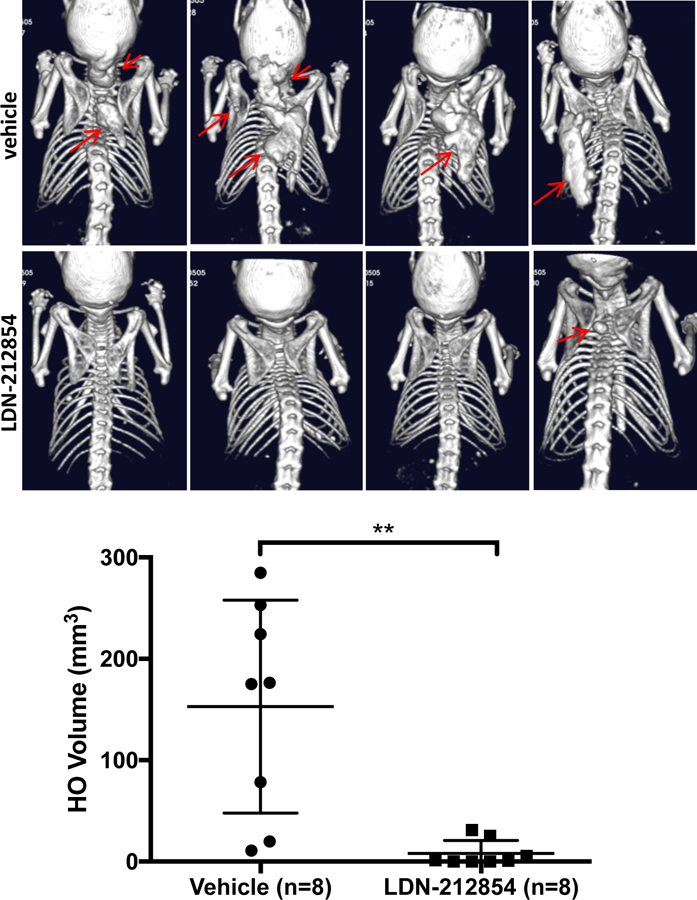
Fibrodysplasia ossificans progressiva (FOP), a congenital heterotopic ossification (HO) syndrome caused by gain-of-function mutations of bone morphogenetic protein (BMP) type I receptor ACVR1, manifests with progressive ossification of skeletal muscles, tendons, ligaments, and joints. In this disease, HO can occur in discrete flares, often triggered by injury or inflammation, or may progress incrementally without identified triggers. Mice harboring an Acvr1R206H knock-in allele recapitulate the phenotypic spectrum of FOP, including injury-responsive intramuscular HO and spontaneous articular, tendon, and ligament ossification. The cells that drive HO in these diverse tissues can be compartmentalized into two lineages: an Scx+ tendon-derived progenitor that mediates endochondral HO of ligaments and joints without exogenous injury, and a muscle-resident interstitial Mx1+ population that mediates intramuscular, injury-dependent endochondral HO. Expression of Acvr1R206H in either lineage confers aberrant gain of BMP signaling and chondrogenic differentiation in response to activin A and gives rise to mutation-expressing hypertrophic chondrocytes in HO lesions. Compared to Acvr1R206H, expression of the man-made, ligand-independent ACVR1Q207D mutation accelerates and increases the penetrance of all observed phenotypes, but does not abrogate the need for antecedent injury in muscle HO, demonstrating the need for an injury factor in addition to enhanced BMP signaling. Both injury-dependent intramuscular and spontaneous ligament HO in Acvr1R206H knock-in mice were effectively controlled by the selective ACVR1 inhibitor LDN-212854. Thus, diverse phenotypes of HO found in FOP are rooted in cell-autonomous effects of dysregulated ACVR1 signaling in nonoverlapping tissue-resident progenitor pools that may be addressed by systemic therapy or by modulating injury-mediated factors involved in their local recruitment.
Copyright © 2016, American Association for the Advancement of Science.
Conflict of interest statement
Figures
References
-
- Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Brown MA, Kaplan FS, A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 38, 525–527 (2006). - PubMed
-
- Kaplan FS, Xu M, Seemann P, Connor JM, Glaser DL, Carroll L, Delai P, Fastnacht-Urban E, Forman SJ, Gillessen-Kaesbach G, Hoover-Fong J, Koster B, Pauli RM, Reardon W, Zaidi SA, Zasloff M, Morhart R, Mundlos S, Groppe J, Shore EM, Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Human mutation 30, 379–390 (2009). - PMC - PubMed
-
- Fukuda T, Kohda M, Kanomata K, Nojima J, Nakamura A, Kamizono J, Noguchi Y, Iwakiri K, Kondo T, Kurose J, Endo K, Awakura T, Fukushi J, Nakashima Y, Chiyonobu T, Kawara A, Nishida Y, Wada I, Akita M, Komori T, Nakayama K, Nanba A, Maruki Y, Yoda T, Tomoda H, Yu PB, Shore EM, Kaplan FS, Miyazono K, Matsuoka M, Ikebuchi K, Ohtake A, Oda H, Jimi E, Owan I, Okazaki Y, Katagiri T, Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J Biol Chem 284, 7149–7156 (2009). - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
