

Enhanced fold recognition using efficient short fragment clustering
- PMID: 27882309
- PMCID: PMC5117261
Enhanced fold recognition using efficient short fragment clustering
Abstract
The main structure aligner in the CCP4 Software Suite, SSM (Secondary Structure Matching) has a limited applicability on the intermediate stages of the structure solution process, when the secondary structure cannot be reliably computed due to structural incompleteness or a fragmented mainchain. In this study, we describe a new algorithm for the alignment and comparison of protein structures in CCP4, which was designed to overcome SSM's limitations but retain its quality and speed. The new algorithm, named GESAMT (General Efficient Structural Alignment of Macromolecular Targets), employs the old idea of deriving the global structure similarity from a promising set of locally similar short fragments, but uses a few technical solutions that make it considerably faster. A comparative sensitivity and selectivity analysis revealed an unexpected significant improvement in the fold recognition properties of the new algorithm, which also makes it useful for applications in the structural bioinformatics domain. The new tool is included in the CCP4 Software Suite starting from version 6.3.
Conflict of interest statement
The author declares no conficts of interest.
Figures
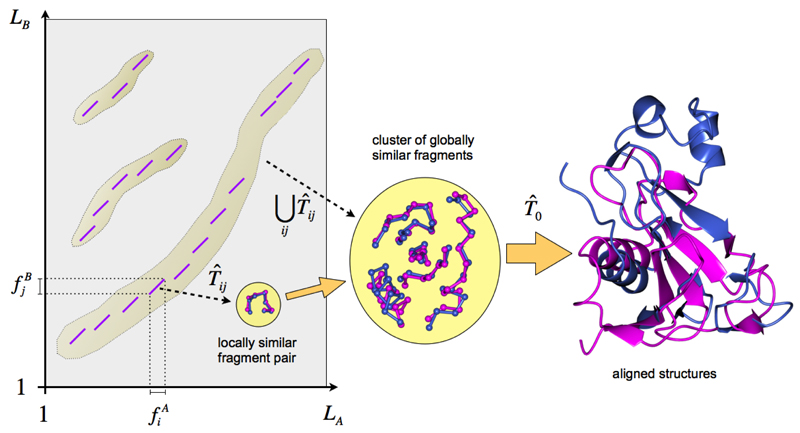
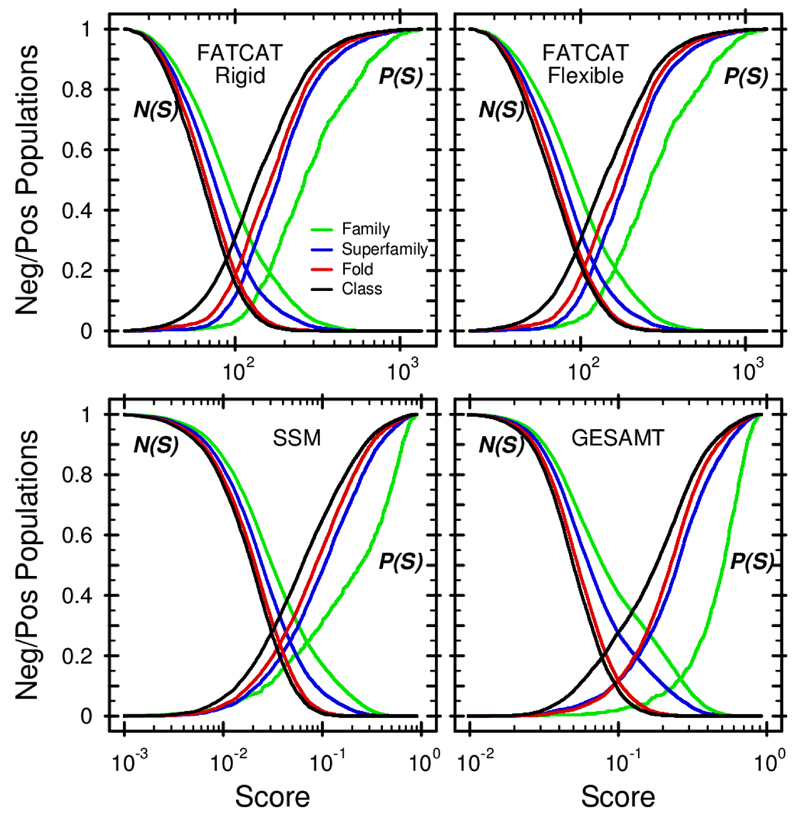
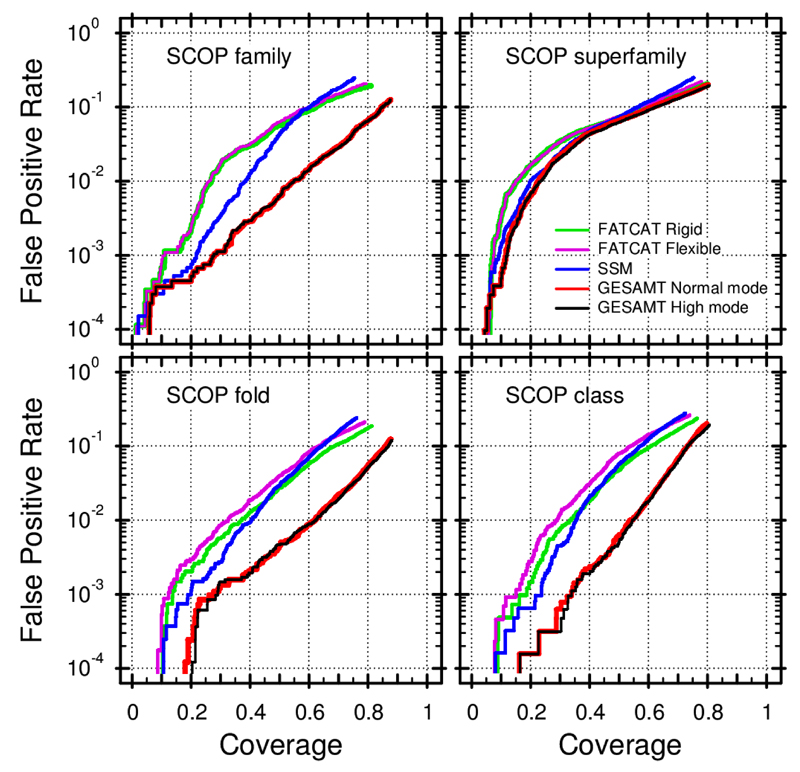
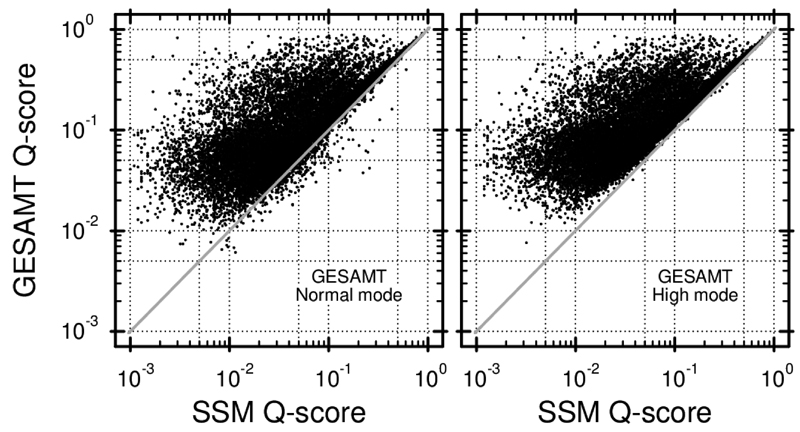
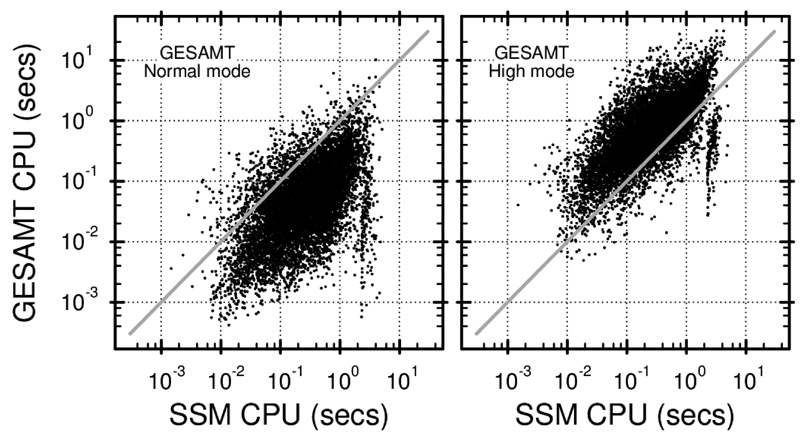
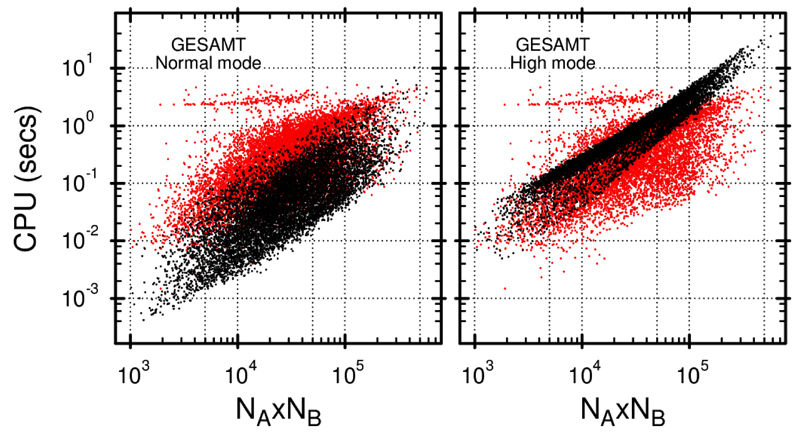
Similar articles
-
CCP4 Cloud for structure determination and project management in macromolecular crystallography.Acta Crystallogr D Struct Biol. 2022 Sep 1;78(Pt 9):1079-1089. doi: 10.1107/S2059798322007987. Epub 2022 Aug 30. Acta Crystallogr D Struct Biol. 2022. PMID: 36048148 Free PMC article.
-
The CCP4 suite: integrative software for macromolecular crystallography.Acta Crystallogr D Struct Biol. 2023 Jun 1;79(Pt 6):449-461. doi: 10.1107/S2059798323003595. Epub 2023 May 30. Acta Crystallogr D Struct Biol. 2023. PMID: 37259835 Free PMC article.
-
UQlust: combining profile hashing with linear-time ranking for efficient clustering and analysis of big macromolecular data.BMC Bioinformatics. 2016 Dec 28;17(1):546. doi: 10.1186/s12859-016-1381-2. BMC Bioinformatics. 2016. PMID: 28031034 Free PMC article.
-
Folic acid supplementation and malaria susceptibility and severity among people taking antifolate antimalarial drugs in endemic areas.Cochrane Database Syst Rev. 2022 Feb 1;2(2022):CD014217. doi: 10.1002/14651858.CD014217. Cochrane Database Syst Rev. 2022. PMID: 36321557 Free PMC article.
-
Ongoing developments in CCP4 for high-throughput structure determination.Acta Crystallogr D Biol Crystallogr. 2002 Nov;58(Pt 11):1929-36. doi: 10.1107/s0907444902016116. Epub 2002 Oct 21. Acta Crystallogr D Biol Crystallogr. 2002. PMID: 12393924 Review.
Cited by
-
Crystal structure of histone-like protein from Streptococcus mutans refined to 1.9 Å resolution.Acta Crystallogr F Struct Biol Commun. 2016 Apr;72(Pt 4):257-62. doi: 10.1107/S2053230X1600217X. Epub 2016 Mar 16. Acta Crystallogr F Struct Biol Commun. 2016. PMID: 27050257 Free PMC article.
-
Structural and biochemical characterization of a novel aminopeptidase from human intestine.J Biol Chem. 2015 May 1;290(18):11321-36. doi: 10.1074/jbc.M114.628149. Epub 2015 Mar 9. J Biol Chem. 2015. PMID: 25752612 Free PMC article.
-
Investigation of how gate residues in the main channel affect the catalytic activity of Scytalidium thermophilum catalase.Acta Crystallogr D Struct Biol. 2024 Feb 1;80(Pt 2):101-112. doi: 10.1107/S2059798323011063. Epub 2024 Jan 24. Acta Crystallogr D Struct Biol. 2024. PMID: 38265876 Free PMC article.
-
Conformational changes in the yeast mitochondrial ABC transporter Atm1 during the transport cycle.Sci Adv. 2021 Dec 24;7(52):eabk2392. doi: 10.1126/sciadv.abk2392. Epub 2021 Dec 22. Sci Adv. 2021. PMID: 34936443 Free PMC article.
-
Concerted removal of the Erb1-Ytm1 complex in ribosome biogenesis relies on an elaborate interface.Nucleic Acids Res. 2016 Jan 29;44(2):926-39. doi: 10.1093/nar/gkv1365. Epub 2015 Dec 10. Nucleic Acids Res. 2016. PMID: 26657628 Free PMC article.
References
-
- Friedberg I, Harder T, Kolodny R, Sitbon E, Li Z, Godzik A. Using an alignment of fragmented strings for comparing protein structures. Bioinformatics. 2007;23:219–224. - PubMed
-
- Gerstein M, Levitt M. Using iterative dynamic programming to obtain accurate pairwise and multiple alignments of protein structures. Proceedings of the Fourth International Conference on Intelligent Systems in Molecular Biology; Menlo Park, CA: AAAI Press; 1996. pp. 59–67. - PubMed
-
- Guerra C, Istrail S. Mathematical methods for protein structure analysis and design: Advanced Lectures. Berlin: Springer Verlag; 2000.
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources