

Hyperglycaemia induces metabolic dysfunction and glycogen accumulation in pancreatic β-cells
- PMID: 27882918
- PMCID: PMC5123088
- DOI: 10.1038/ncomms13496
Hyperglycaemia induces metabolic dysfunction and glycogen accumulation in pancreatic β-cells
Abstract
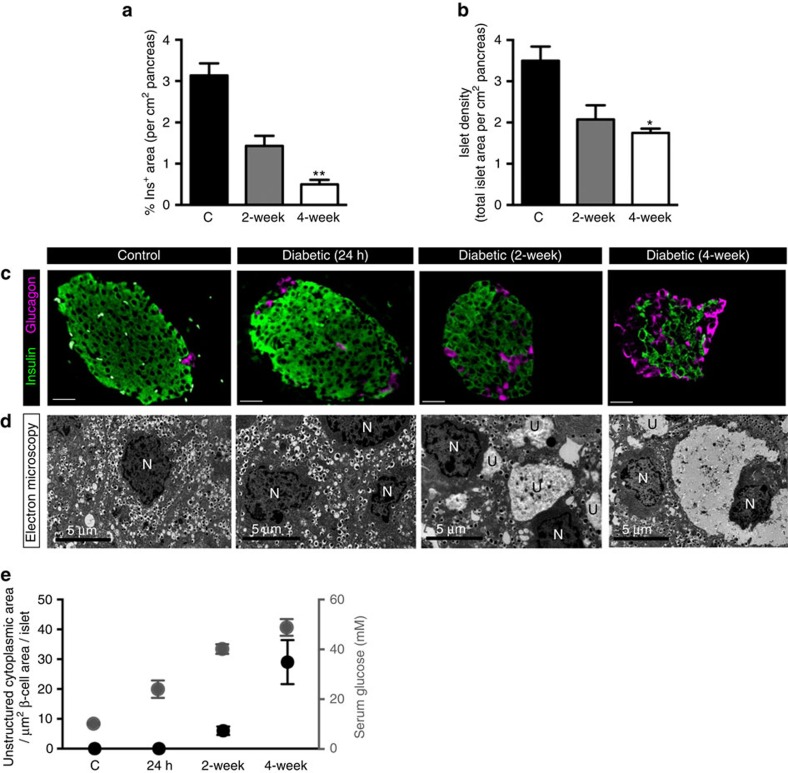
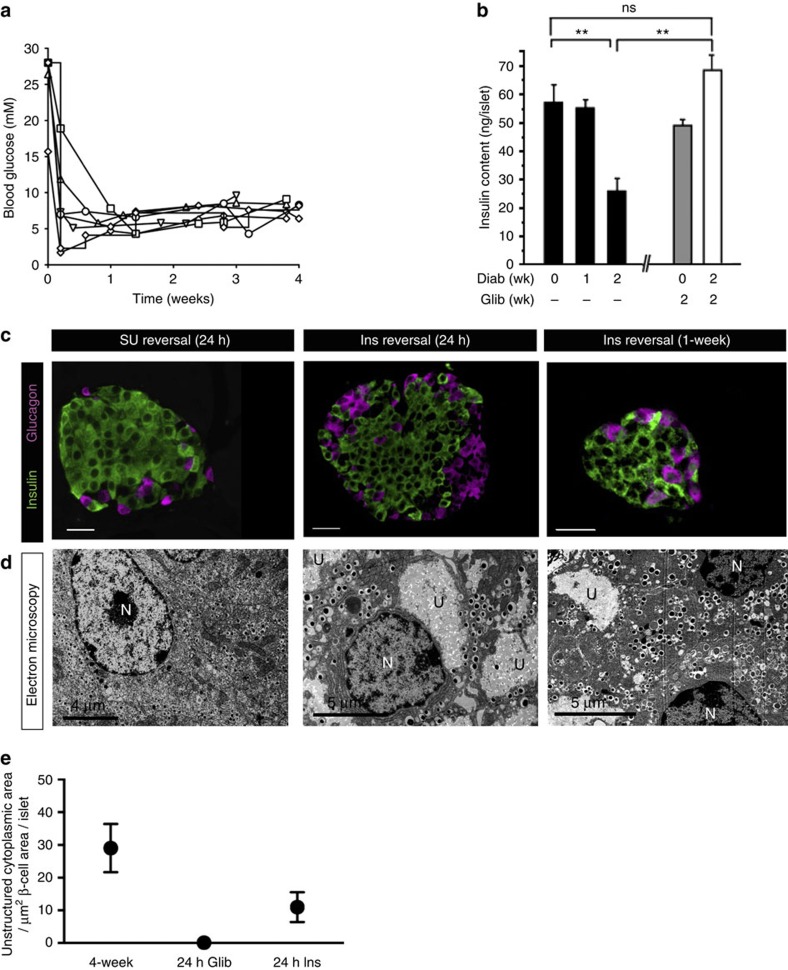
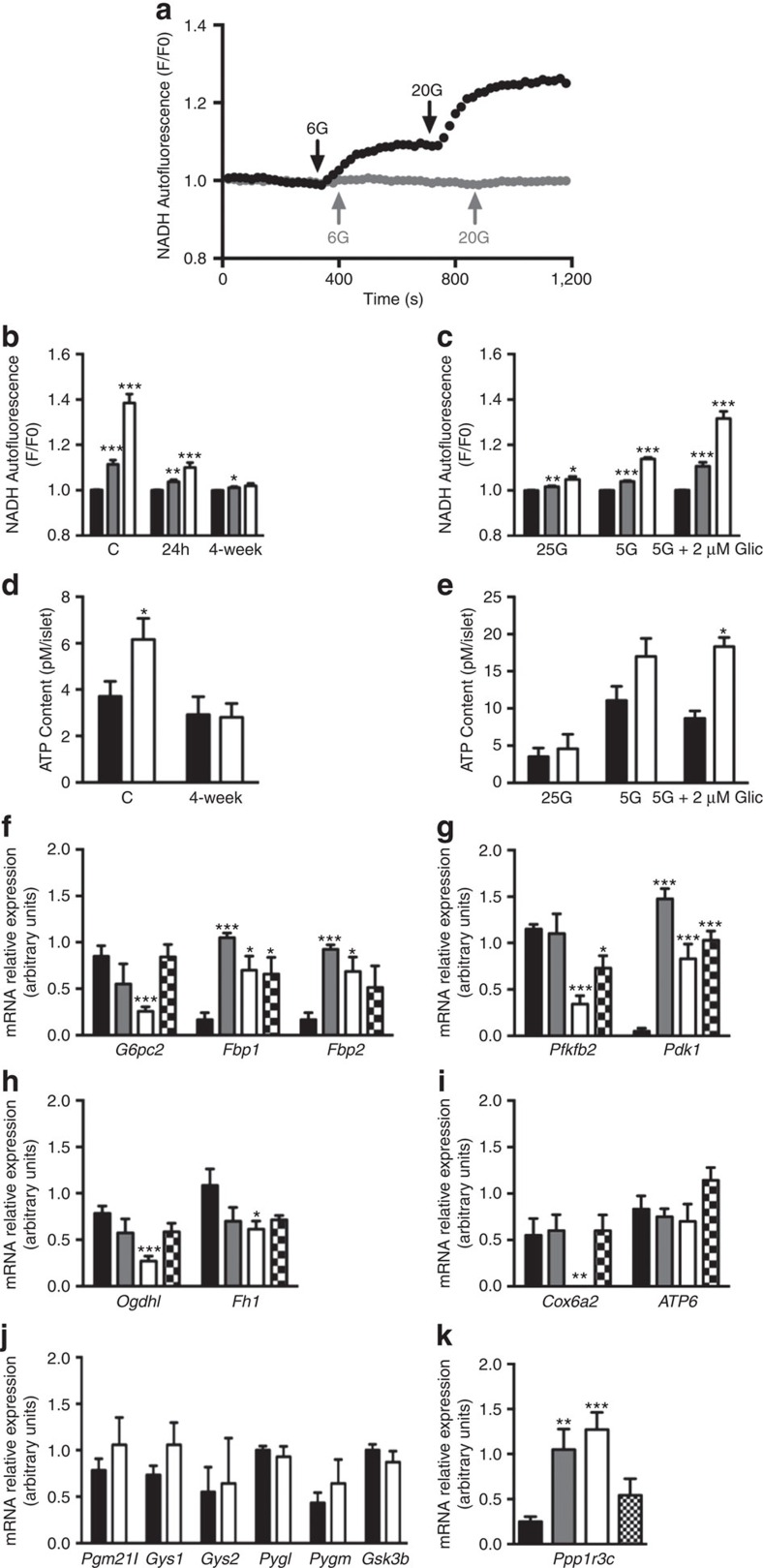
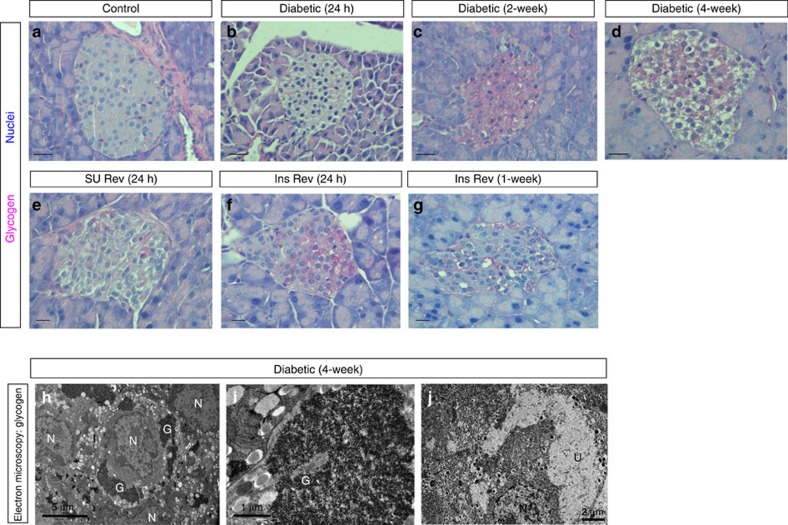
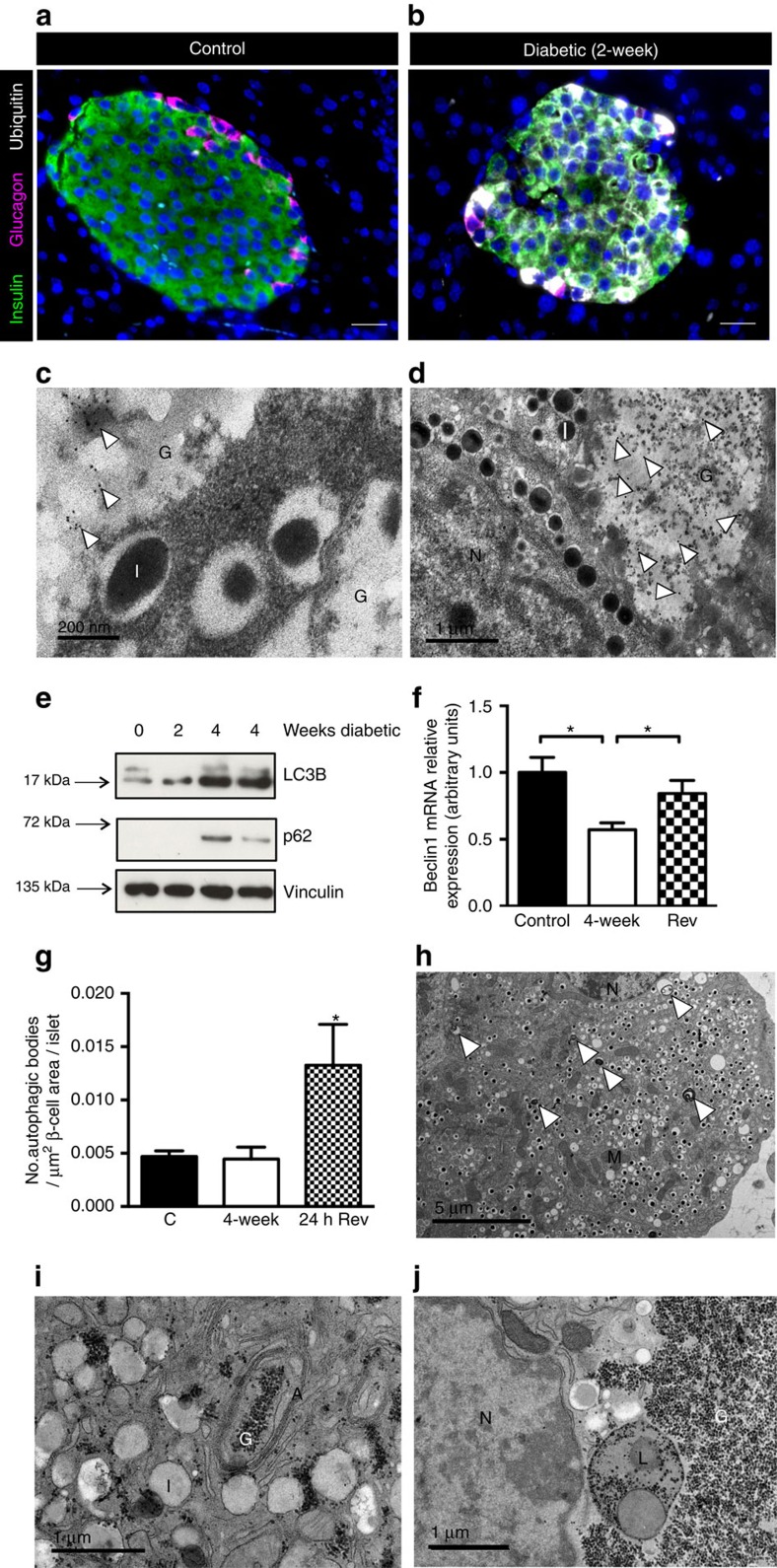
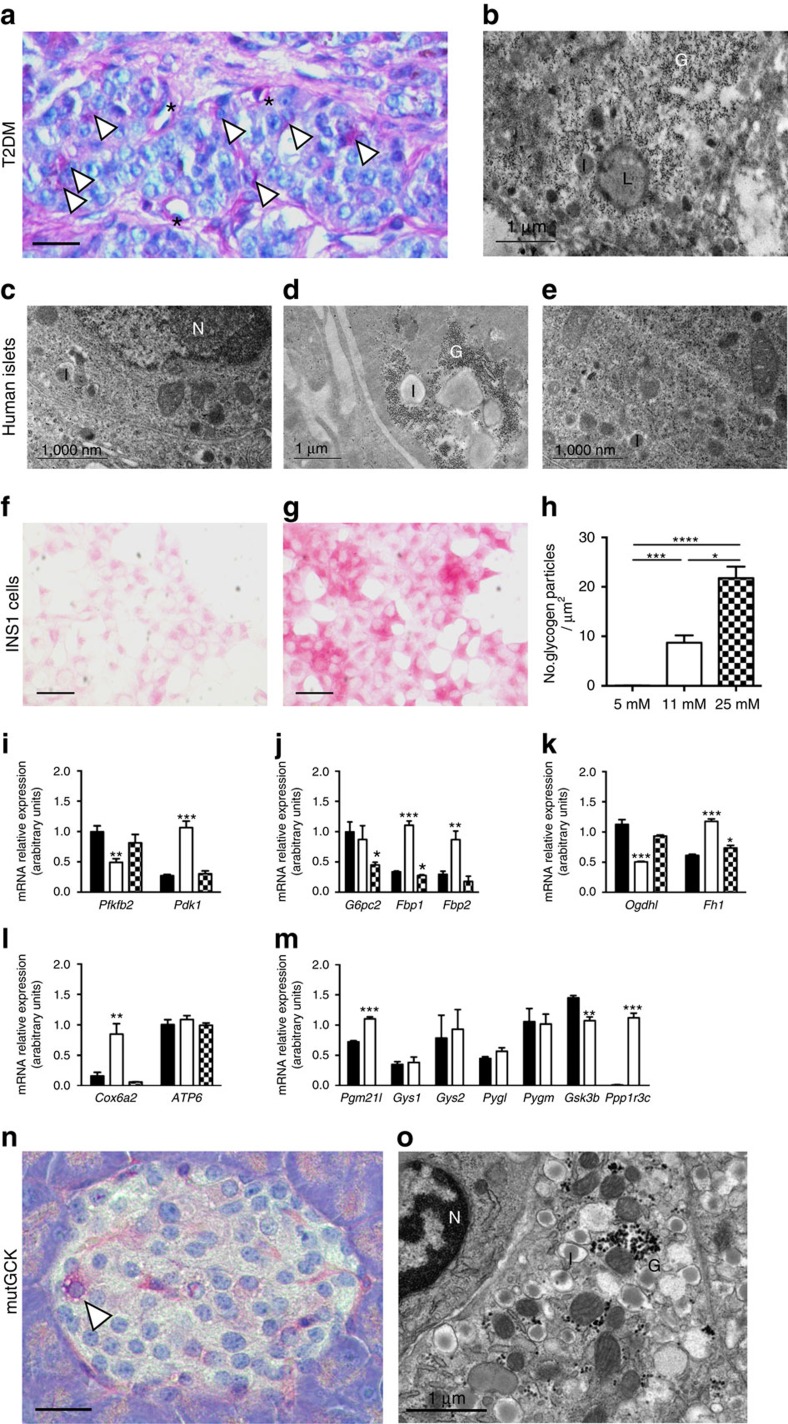
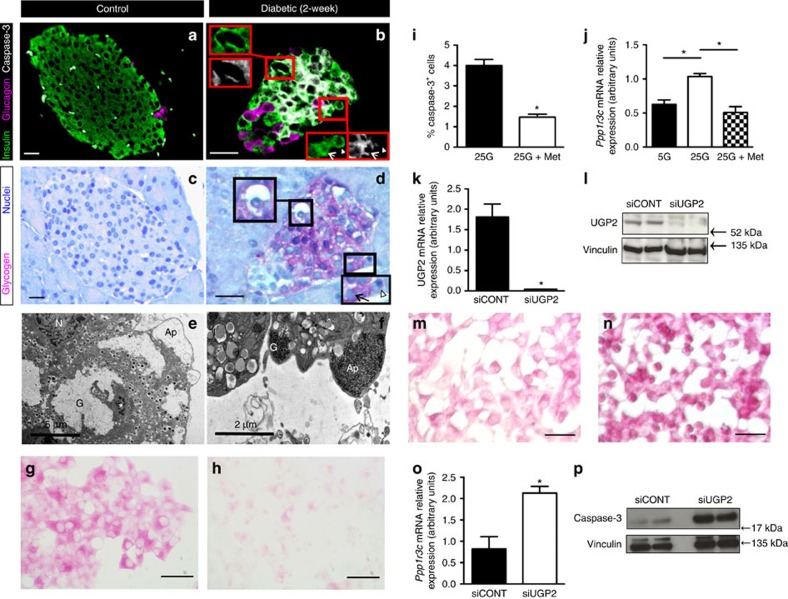
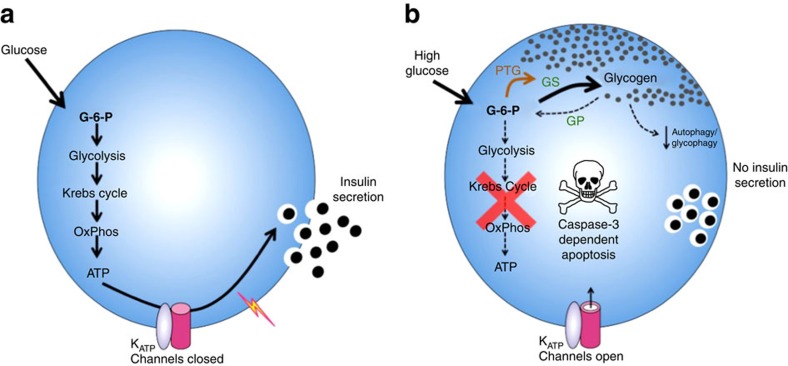
Insulin secretion from pancreatic β-cells is impaired in all forms of diabetes. The resultant hyperglycaemia has deleterious effects on many tissues, including β-cells. Here we show that chronic hyperglycaemia impairs glucose metabolism and alters expression of metabolic genes in pancreatic islets. In a mouse model of human neonatal diabetes, hyperglycaemia results in marked glycogen accumulation, and increased apoptosis in β-cells. Sulphonylurea therapy rapidly normalizes blood glucose levels, dissipates glycogen stores, increases autophagy and restores β-cell metabolism. Insulin therapy has the same effect but with slower kinetics. Similar changes are observed in mice expressing an activating glucokinase mutation, in in vitro models of hyperglycaemia, and in islets from type-2 diabetic patients. Altered β-cell metabolism may underlie both the progressive impairment of insulin secretion and reduced β-cell mass in diabetes.
Figures
References
-
- Jonas J. C. et al.. Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. J. Biol. Chem. 274, 14112–14121 (1999). - PubMed
-
- Henquin J. C. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49, 1751–1760 (2000). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
