

Inferring Cell-State Transition Dynamics from Lineage Trees and Endpoint Single-Cell Measurements
- PMID: 27883889
- PMCID: PMC5142829
- DOI: 10.1016/j.cels.2016.10.015
Inferring Cell-State Transition Dynamics from Lineage Trees and Endpoint Single-Cell Measurements
Abstract
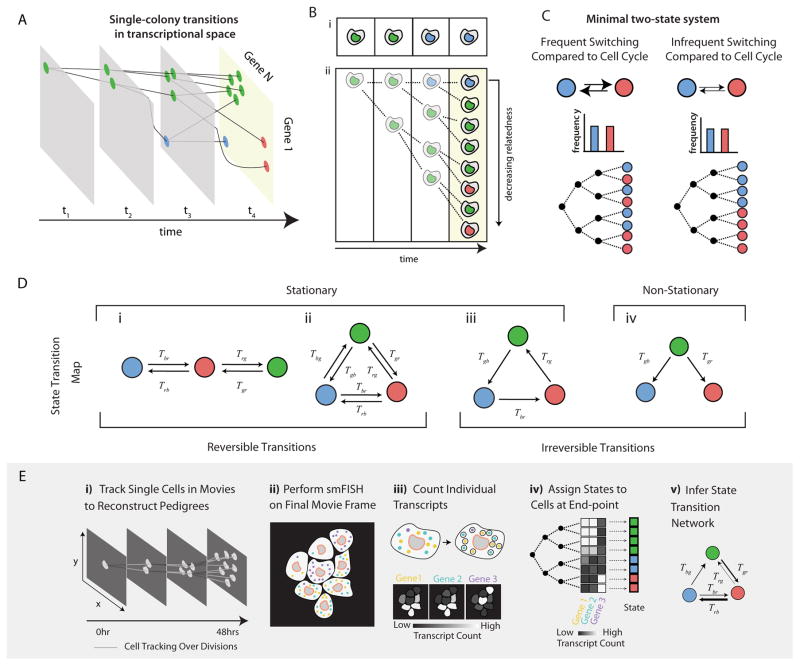
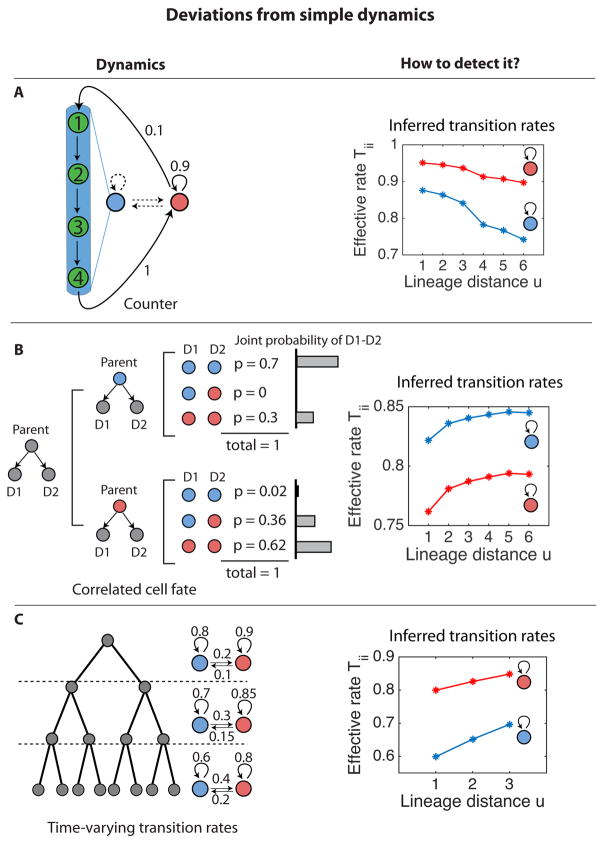
As they proliferate, living cells undergo transitions between specific molecularly and developmentally distinct states. Despite the functional centrality of these transitions in multicellular organisms, it has remained challenging to determine which transitions occur and at what rates without perturbations and cell engineering. Here, we introduce kin correlation analysis (KCA) and show that quantitative cell-state transition dynamics can be inferred, without direct observation, from the clustering of cell states on pedigrees (lineage trees). Combining KCA with pedigrees obtained from time-lapse imaging and endpoint single-molecule RNA-fluorescence in situ hybridization (RNA-FISH) measurements of gene expression, we determined the cell-state transition network of mouse embryonic stem (ES) cells. This analysis revealed that mouse ES cells exhibit stochastic and reversible transitions along a linear chain of states ranging from 2C-like to epiblast-like. Our approach is broadly applicable and may be applied to systems with irreversible transitions and non-stationary dynamics, such as in cancer and development.
Keywords: cell state transition; dynamics; heterogeneity; inference; lineage; single cell; single-molecule FISH; stem cells; stochasticity; time-lapse microscopy.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures



Similar articles
-
Synthetic recording and in situ readout of lineage information in single cells.Nature. 2017 Jan 5;541(7635):107-111. doi: 10.1038/nature20777. Epub 2016 Nov 21. Nature. 2017. PMID: 27869821 Free PMC article.
-
Single Cell Analysis Reveals Concomitant Transcription of Pluripotent and Lineage Markers During the Early Steps of Differentiation of Embryonic Stem Cells.Stem Cells. 2015 Oct;33(10):2949-60. doi: 10.1002/stem.2108. Stem Cells. 2015. PMID: 26184691
-
Analysis of Cell Lineage Trees by Exact Bayesian Inference Identifies Negative Autoregulation of Nanog in Mouse Embryonic Stem Cells.Cell Syst. 2016 Nov 23;3(5):480-490.e13. doi: 10.1016/j.cels.2016.11.001. Cell Syst. 2016. PMID: 27883891
-
The role of pluripotency gene regulatory network components in mediating transitions between pluripotent cell states.Curr Opin Genet Dev. 2013 Oct;23(5):504-11. doi: 10.1016/j.gde.2013.06.003. Epub 2013 Aug 7. Curr Opin Genet Dev. 2013. PMID: 23932125 Free PMC article. Review.
-
Pluripotency Deconstructed.Dev Growth Differ. 2018 Jan;60(1):44-52. doi: 10.1111/dgd.12419. Epub 2018 Jan 23. Dev Growth Differ. 2018. PMID: 29359419 Review.
Cited by
-
A stochastic epigenetic switch controls the dynamics of T-cell lineage commitment.Elife. 2018 Nov 20;7:e37851. doi: 10.7554/eLife.37851. Elife. 2018. PMID: 30457103 Free PMC article.
-
Stem Cell Differentiation as a Non-Markov Stochastic Process.Cell Syst. 2017 Sep 27;5(3):268-282.e7. doi: 10.1016/j.cels.2017.08.009. Cell Syst. 2017. PMID: 28957659 Free PMC article.
-
Gene expression at a single-molecule level: implications for myelodysplastic syndromes and acute myeloid leukemia.Blood. 2021 Aug 26;138(8):625-636. doi: 10.1182/blood.2019004261. Blood. 2021. PMID: 34436525 Free PMC article. Review.
-
Multiplex Protein Imaging through PACIFIC: Photoactive Immunofluorescence with Iterative Cleavage.ACS Bio Med Chem Au. 2023 Apr 28;3(3):283-294. doi: 10.1021/acsbiomedchemau.3c00018. eCollection 2023 Jun 21. ACS Bio Med Chem Au. 2023. PMID: 37363079 Free PMC article.
-
Integrated spatial genomics reveals global architecture of single nuclei.Nature. 2021 Feb;590(7845):344-350. doi: 10.1038/s41586-020-03126-2. Epub 2021 Jan 27. Nature. 2021. PMID: 33505024 Free PMC article.
References
-
- Chambers I, Silva J, Colby D, Nichols J, Nijmeijer B, Robertson M, Vrana J, Jones K, Grotewold L, Smith A. Nanog safeguards pluripotency and mediates germline development. Nature. 2007;450:1230–1234. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
