

variancePartition: interpreting drivers of variation in complex gene expression studies
- PMID: 27884101
- PMCID: PMC5123296
- DOI: 10.1186/s12859-016-1323-z
variancePartition: interpreting drivers of variation in complex gene expression studies
Abstract
Background: As large-scale studies of gene expression with multiple sources of biological and technical variation become widely adopted, characterizing these drivers of variation becomes essential to understanding disease biology and regulatory genetics.
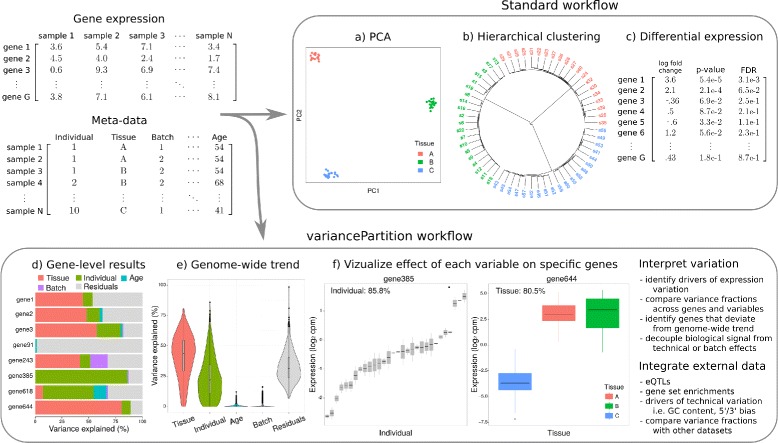
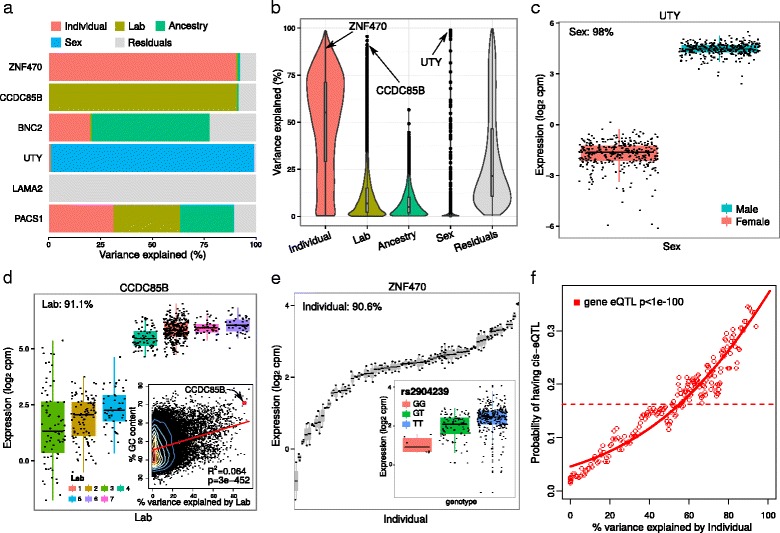
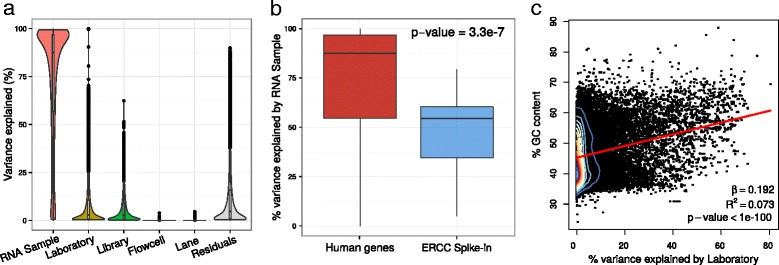
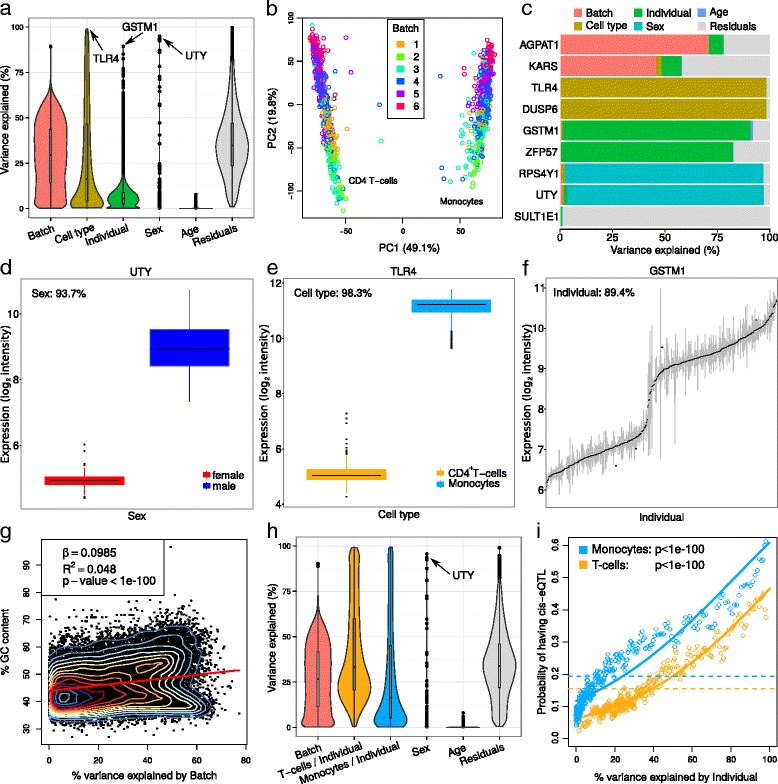
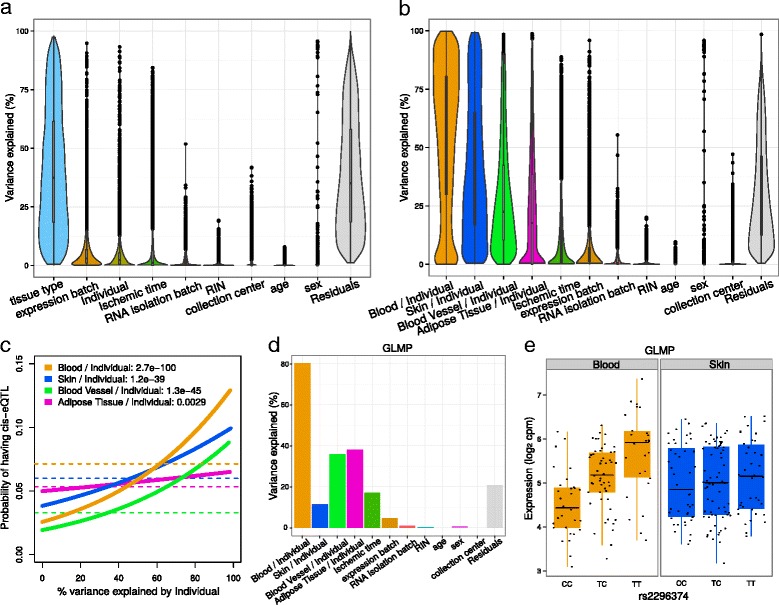
Results: We describe a statistical and visualization framework, variancePartition, to prioritize drivers of variation based on a genome-wide summary, and identify genes that deviate from the genome-wide trend. Using a linear mixed model, variancePartition quantifies variation in each expression trait attributable to differences in disease status, sex, cell or tissue type, ancestry, genetic background, experimental stimulus, or technical variables. Analysis of four large-scale transcriptome profiling datasets illustrates that variancePartition recovers striking patterns of biological and technical variation that are reproducible across multiple datasets.
Conclusions: Our open source software, variancePartition, enables rapid interpretation of complex gene expression studies as well as other high-throughput genomics assays. variancePartition is available from Bioconductor: http://bioconductor.org/packages/variancePartition .
Keywords: Linear mixed model; RNA-seq; Transcriptome profiling.
Figures
References
-
- Raj T, Rothamel K, Mostafavi S, Ye C, Lee MMN, Replogle JM, Feng T, Asinovski N, Frohlich I, Imboywa S, Von Korff A, Okada Y, Patsopoulos NA, Davis S, McCabe C, Paik H-I, Srivastava GP, Raychaudhuri S, Hafler DA, Koller D, Regev A, Hacohen N, Mathis D, Benoist C, Stranger BE, De Jager PL. Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science. 2014;344(6183):519–23. doi: 10.1126/science.1249547. - DOI - PMC - PubMed
-
- Ramasamy A, Trabzuni D, Guelfi S, Varghese V, Smith C, Walker R, De T, Hardy J, Ryten M, Trabzuni D, Guelfi S, Weale ME, Ramasamy A, Forabosco P, Smith C, Walker R, Arepalli S, Cookson MR, Dillman A, Gibbs JR, Hernandez DG, Nalls MA, Singleton AB, Traynor B, van der Brug M, Ferrucci L, Johnson R, Zielke R, Longo DL, Troncoso J, Zonderman A, Coin L, de Silva R, Cookson MR, Singleton AB, Hardy J, Ryten M, Weale ME. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat Neurosci. 2014;17(10):1418–28. doi: 10.1038/nn.3801. - DOI - PMC - PubMed
-
- Lee MN, Ye C, Villani AC, Raj T, Li W, Eisenhaure TM, Imboywa SH, Chipendo PI, Ran FA, Slowikowski K, Ward LD, Raddassi K, McCabe C, Lee MH, Frohlich IY, Hafler D. a, Kellis M, Raychaudhuri S, Zhang F, Stranger BE, Benoist CO, De Jager PL, Regev A, Hacohen N. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science. 2014;343(6175):1246980. doi: 10.1126/science.1246980. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
