

Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary
- PMID: 27887642
- PMCID: PMC5124306
- DOI: 10.1186/s13059-016-1108-8
Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary
Erratum in
-
Erratum to: Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary.Genome Biol. 2016 Dec 19;17(1):262. doi: 10.1186/s13059-016-1132-8. Genome Biol. 2016. PMID: 27993146 Free PMC article. No abstract available.
Abstract
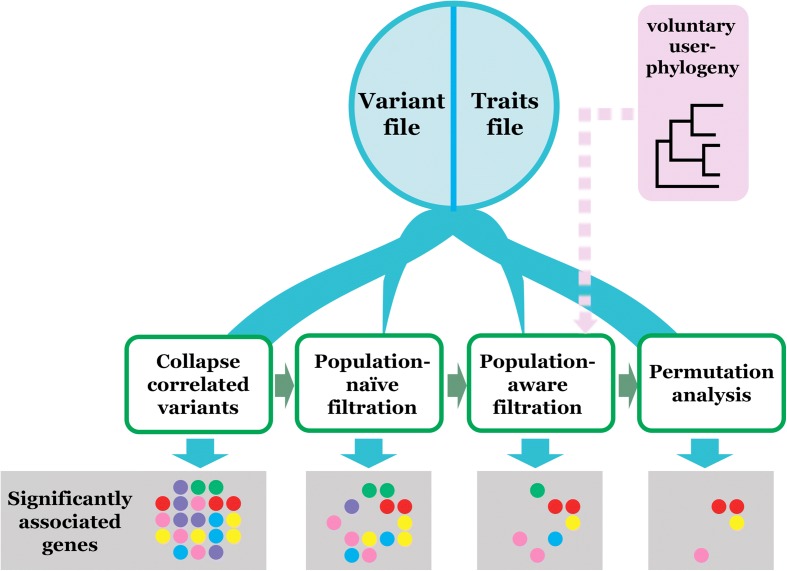
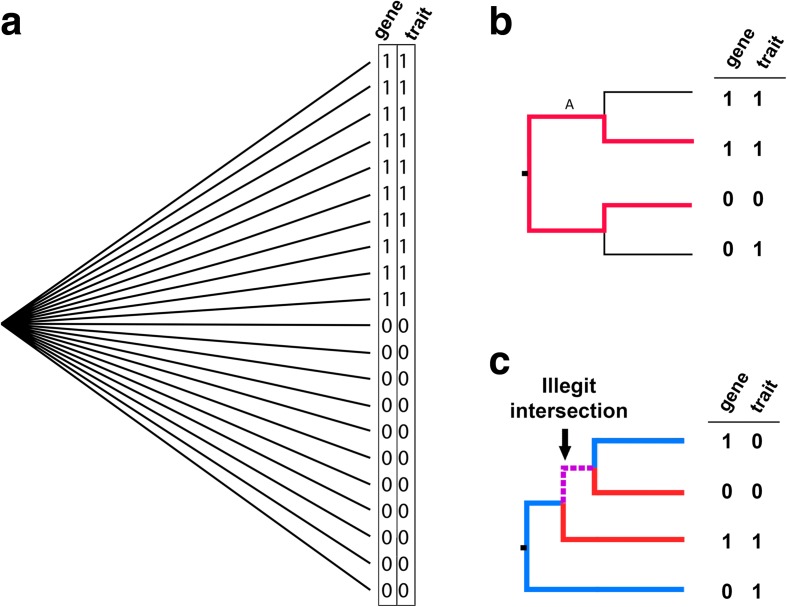
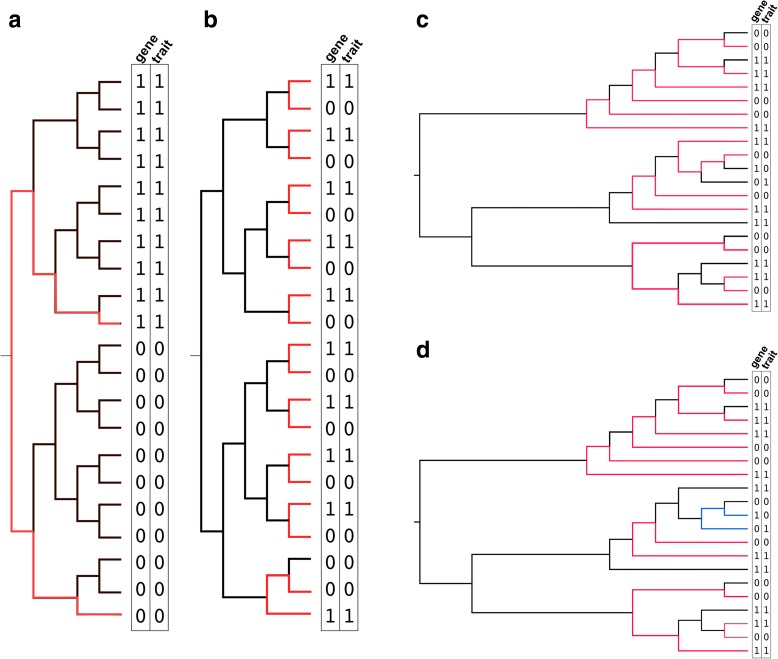
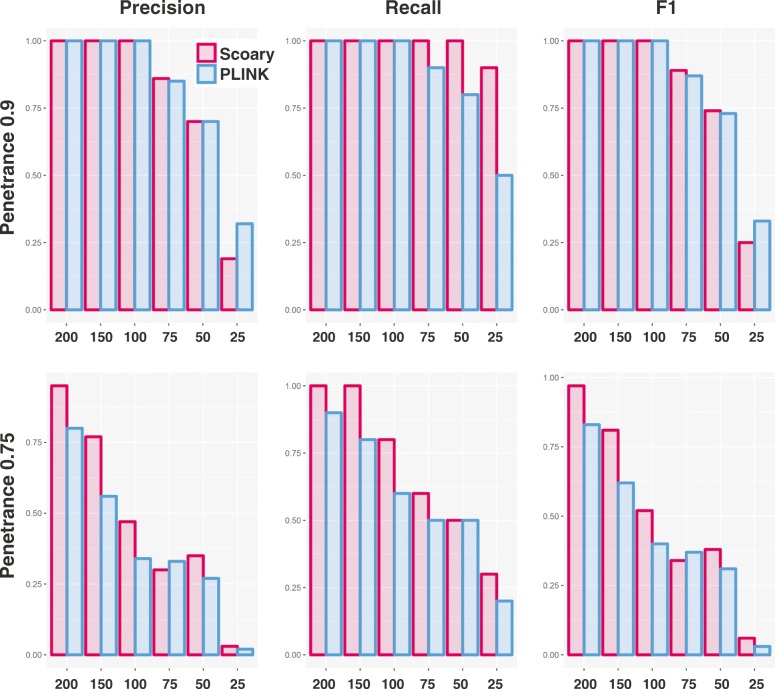
Genome-wide association studies (GWAS) have become indispensable in human medicine and genomics, but very few have been carried out on bacteria. Here we introduce Scoary, an ultra-fast, easy-to-use, and widely applicable software tool that scores the components of the pan-genome for associations to observed phenotypic traits while accounting for population stratification, with minimal assumptions about evolutionary processes. We call our approach pan-GWAS to distinguish it from traditional, single nucleotide polymorphism (SNP)-based GWAS. Scoary is implemented in Python and is available under an open source GPLv3 license at https://github.com/AdmiralenOla/Scoary .
Keywords: Accessory genome; Annotation; Association; Bacteria; Genome-wide association studies (GWAS); Genomics; Next-generation sequencing (NGS); Pan-genome; Prokaryote; Python; Whole-genome sequencing (WGS).
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
