

Involvement of inflammation and its related microRNAs in hepatocellular carcinoma
- PMID: 27888618
- PMCID: PMC5400654
- DOI: 10.18632/oncotarget.13530
Involvement of inflammation and its related microRNAs in hepatocellular carcinoma
Abstract
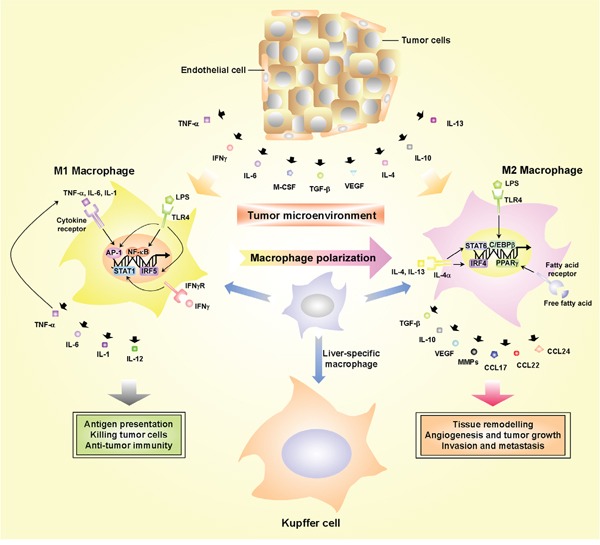
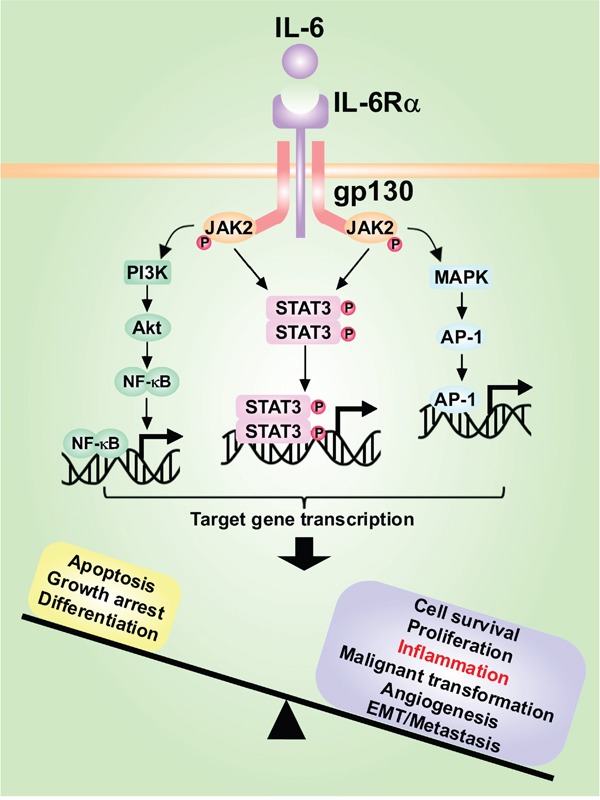
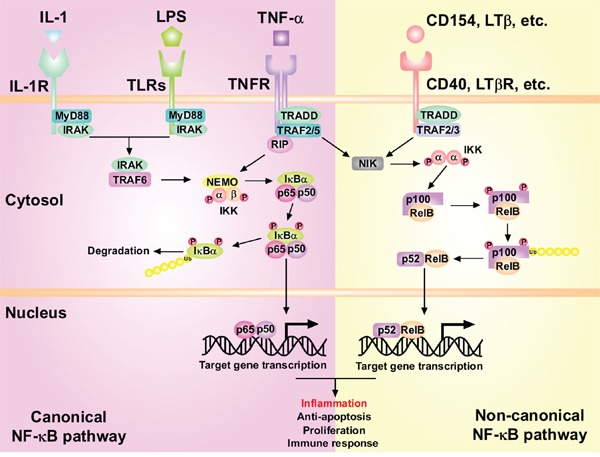
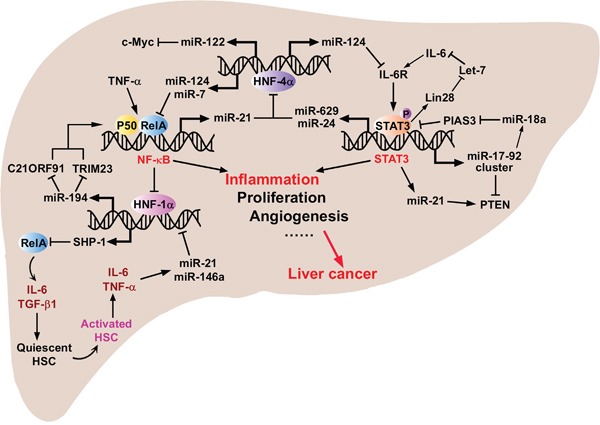
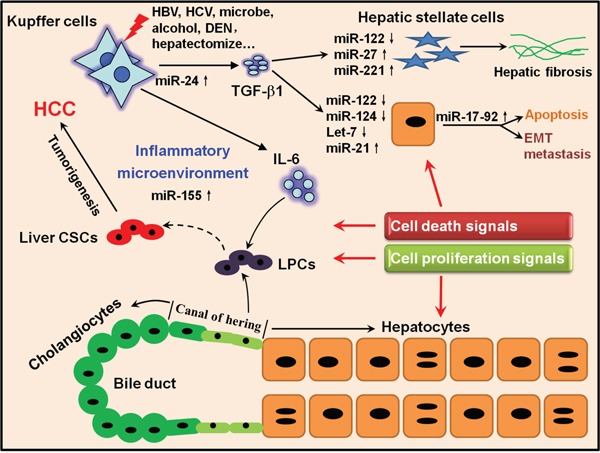
Hepatocellular carcinoma (HCC) is the fifth most commonly diagnosed type of cancer. The tumor inflammatory microenvironment regulates almost every step towards liver tumorigenesis and subsequent progression, and regulation of the inflammation-related signaling pathways, cytokines, chemokines and non-coding RNAs influences the proliferation, migration and metastasis of liver tumor cells. Inflammation fine-tunes the cancer microenvironment to favor epithelial-mesenchymal transition, in which cancer stem cells maintain tumorigenic potential. Emerging evidence points to inflammation-related microRNAs as crucial molecules to integrate the complex cellular and molecular crosstalk during HCC progression. Thus understanding the mechanisms by which inflammation regulates microRNAs might provide novel and admissible strategies for preventing, diagnosing and treating HCC. In this review, we will update three hypotheses of hepatocarcinogenesis and elaborate the most predominant inflammation signaling pathways, i.e. IL-6/STAT3 and NF-κB. We also try to summarize the crucial tumor-promoting and tumor-suppressing microRNAs and detail how they regulate HCC initiation and progression and collaborate with other critical modulators in this review.
Keywords: cancer stem cells; cell signaling; epithelial-mesenchymal transition; inflammation; microRNAs.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Cellular and molecular mechanisms of hepatocellular carcinoma: an update.Arch Toxicol. 2013 Feb;87(2):227-47. doi: 10.1007/s00204-012-0931-2. Epub 2012 Sep 25. Arch Toxicol. 2013. PMID: 23007558 Review.
-
Small but Heavy Role: MicroRNAs in Hepatocellular Carcinoma Progression.Biomed Res Int. 2018 May 22;2018:6784607. doi: 10.1155/2018/6784607. eCollection 2018. Biomed Res Int. 2018. PMID: 29951542 Free PMC article. Review.
-
NF-κB signaling relieves negative regulation by miR-194 in hepatocellular carcinoma by suppressing the transcription factor HNF-1α.Sci Signal. 2015 Jul 28;8(387):ra75. doi: 10.1126/scisignal.aaa8441. Sci Signal. 2015. PMID: 26221053
-
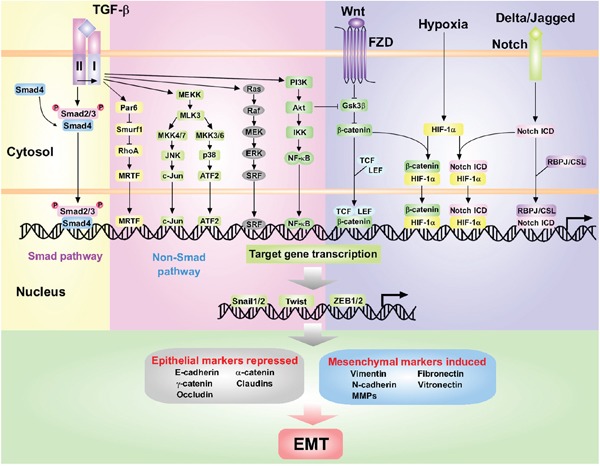
Relationship between epithelial-to-mesenchymal transition and the inflammatory microenvironment of hepatocellular carcinoma.J Exp Clin Cancer Res. 2018 Aug 29;37(1):203. doi: 10.1186/s13046-018-0887-z. J Exp Clin Cancer Res. 2018. PMID: 30157906 Free PMC article. Review.
-
MicroRNA-451: epithelial-mesenchymal transition inhibitor and prognostic biomarker of hepatocelluar carcinoma.Oncotarget. 2015 Jul 30;6(21):18613-30. doi: 10.18632/oncotarget.4317. Oncotarget. 2015. PMID: 26164082 Free PMC article.
Cited by
-
A comprehensive insight into the clinicopathologic significance of miR-144-3p in hepatocellular carcinoma.Onco Targets Ther. 2017 Jul 11;10:3405-3419. doi: 10.2147/OTT.S138143. eCollection 2017. Onco Targets Ther. 2017. PMID: 28744145 Free PMC article.
-
Construction and application of a liver cancer-targeting drug delivery system based on core-shell gold nanocages.Int J Nanomedicine. 2018 Mar 21;13:1773-1789. doi: 10.2147/IJN.S151043. eCollection 2018. Int J Nanomedicine. 2018. PMID: 29606870 Free PMC article.
-
miR-579-3p Controls Hepatocellular Carcinoma Formation by Regulating the Phosphoinositide 3-Kinase-Protein Kinase B Pathway in Chronically Inflamed Liver.Hepatol Commun. 2022 Jun;6(6):1467-1481. doi: 10.1002/hep4.1894. Epub 2022 Feb 8. Hepatol Commun. 2022. PMID: 35132819 Free PMC article.
-
Ferroptosis in Intrahepatic Cholangiocarcinoma: IDH1105GGT Single Nucleotide Polymorphism Is Associated With Its Activation and Better Prognosis.Front Med (Lausanne). 2022 Jul 8;9:886229. doi: 10.3389/fmed.2022.886229. eCollection 2022. Front Med (Lausanne). 2022. PMID: 35872783 Free PMC article.
-
Identifying key regulating miRNAs in hepatocellular carcinomas by an omics' method.Oncotarget. 2017 Oct 17;8(61):103919-103930. doi: 10.18632/oncotarget.21865. eCollection 2017 Nov 28. Oncotarget. 2017. PMID: 29262610 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
