

MUC1-C activates BMI1 in human cancer cells
- PMID: 27893710
- PMCID: PMC5436937
- DOI: 10.1038/onc.2016.439
MUC1-C activates BMI1 in human cancer cells
Abstract
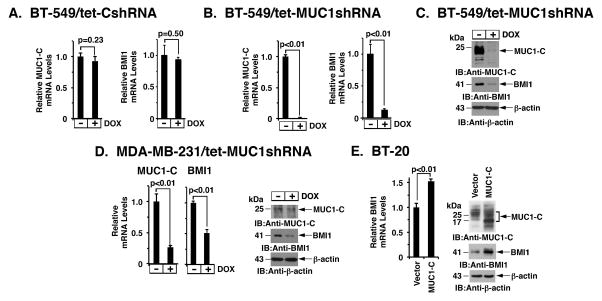
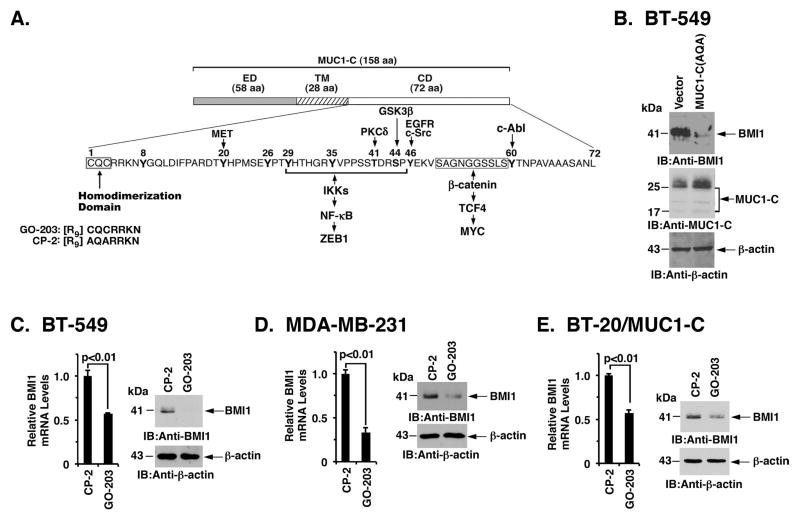
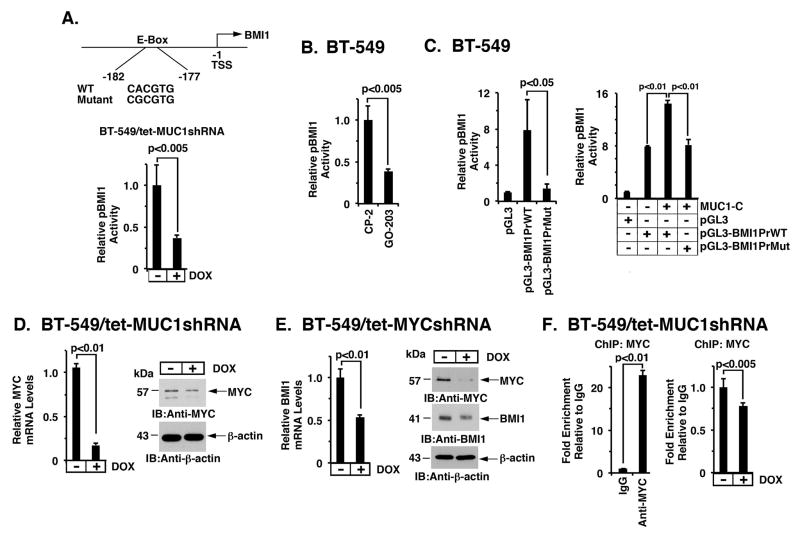
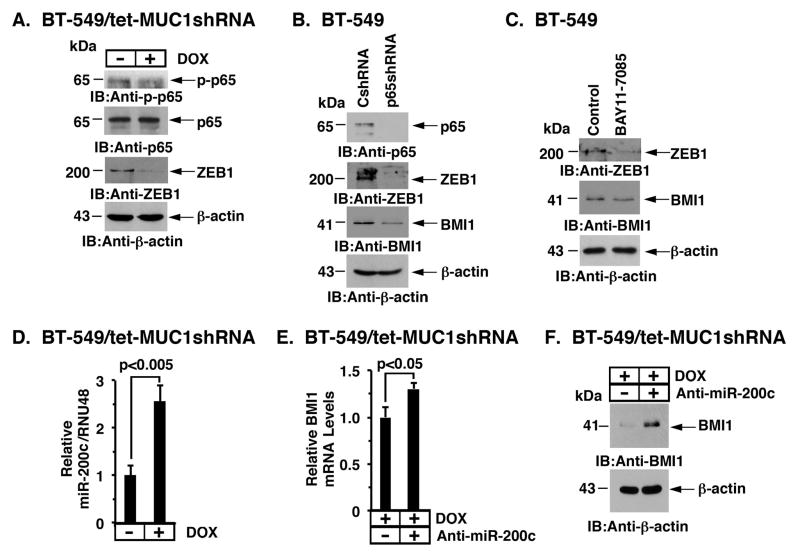
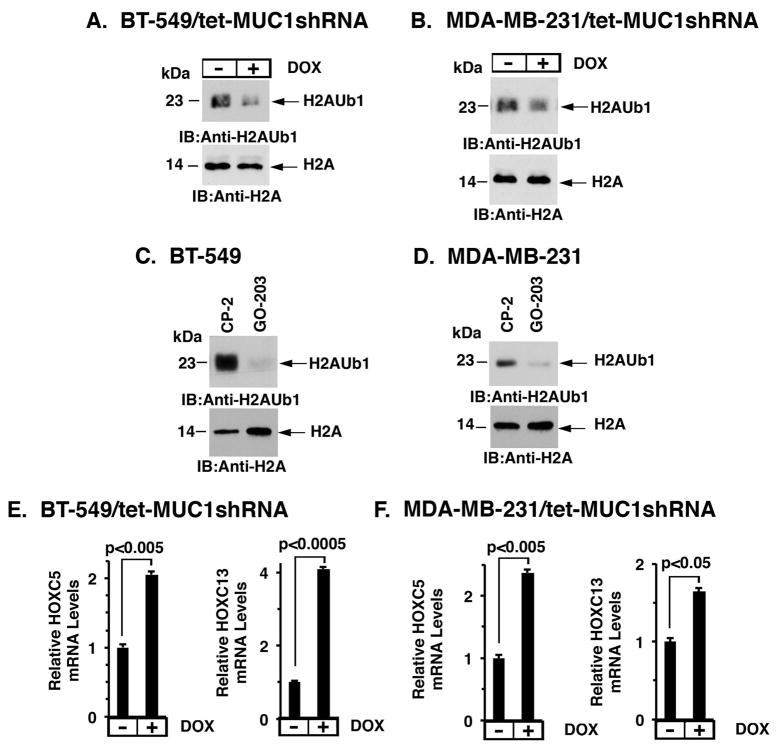
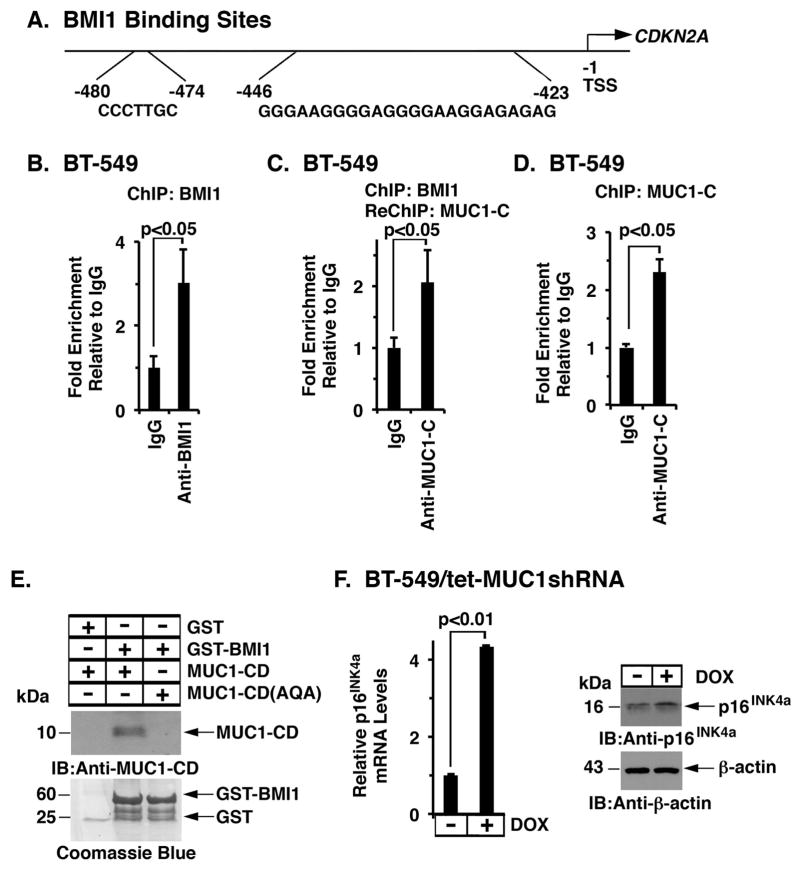
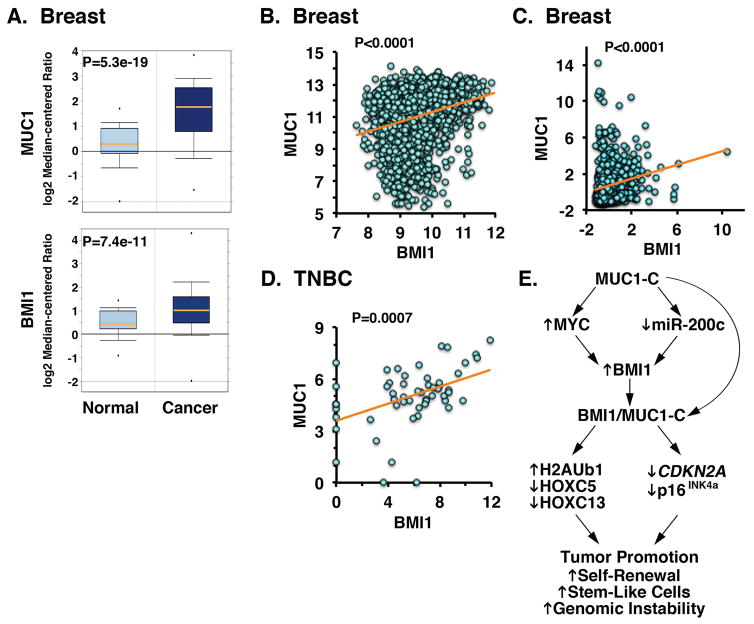
B-cell-specific Moloney murine leukemia virus integration site 1 (BMI1) is a component of the polycomb repressive complex 1 (PRC1) complex that is overexpressed in breast and other cancers, and promotes self-renewal of cancer stem-like cells. The oncogenic mucin 1 (MUC1) C-terminal (MUC1-C) subunit is similarly overexpressed in human carcinoma cells and has been linked to their self-renewal. There is no known relationship between MUC1-C and BMI1 in cancer. The present studies demonstrate that MUC1-C drives BMI1 transcription by a MYC-dependent mechanism in breast and other cancer cells. In addition, we show that MUC1-C blocks miR-200c-mediated downregulation of BMI1 expression. The functional significance of this MUC1-C→︀BMI1 pathway is supported by the demonstration that targeting MUC1-C suppresses BMI1-induced ubiquitylation of H2A and thereby derepresses homeobox HOXC5 and HOXC13 gene expression. Notably, our results further show that MUC1-C binds directly to BMI1 and promotes occupancy of BMI1 on the CDKN2A promoter. In concert with BMI1-induced repression of the p16INK4a tumor suppressor, we found that targeting MUC1-C is associated with induction of p16INK4a expression. In support of these results, analysis of three gene expresssion data sets demonstrated highly significant correlations between MUC1-C and BMI1 in breast cancers. These findings uncover a previously unrecognized role for MUC1-C in driving BMI1 expression and in directly interacting with this stem cell factor, linking MUC1-C with function of the PRC1 in epigenetic gene silencing.
Conflict of interest statement
The authors declare competing financial interests: D.K. holds equity in Genus Oncology and is a consultant to the company. The other authors disclosed no potential conflicts of interest.
Figures
References
-
- Siddique HR, Saleem M. Role of BMI1, a stem cell factor, in cancer recurrence and chemoresistance: preclinical and clinical evidences. Stem Cells. 2012;30:372–8. - PubMed
-
- Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431:873–8. - PubMed
-
- de Napoles M, Mermoud JE, Wakao R, Tang YA, Endoh M, Appanah R, et al. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev Cell. 2004;7:663–76. - PubMed
-
- Cao R, Tsukada Y, Zhang Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell. 2005;20:845–54. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
