

Simulating Next-Generation Sequencing Datasets from Empirical Mutation and Sequencing Models
- PMID: 27893777
- PMCID: PMC5125660
- DOI: 10.1371/journal.pone.0167047
Simulating Next-Generation Sequencing Datasets from Empirical Mutation and Sequencing Models
Abstract
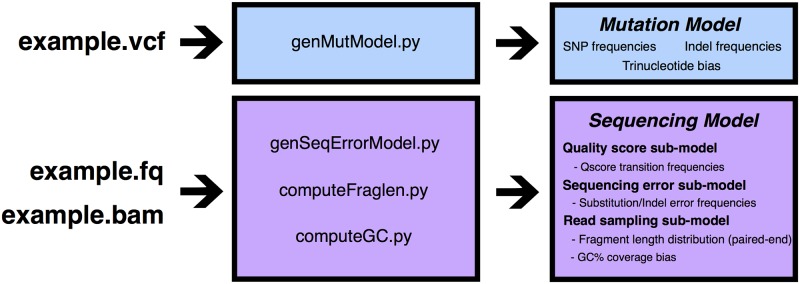
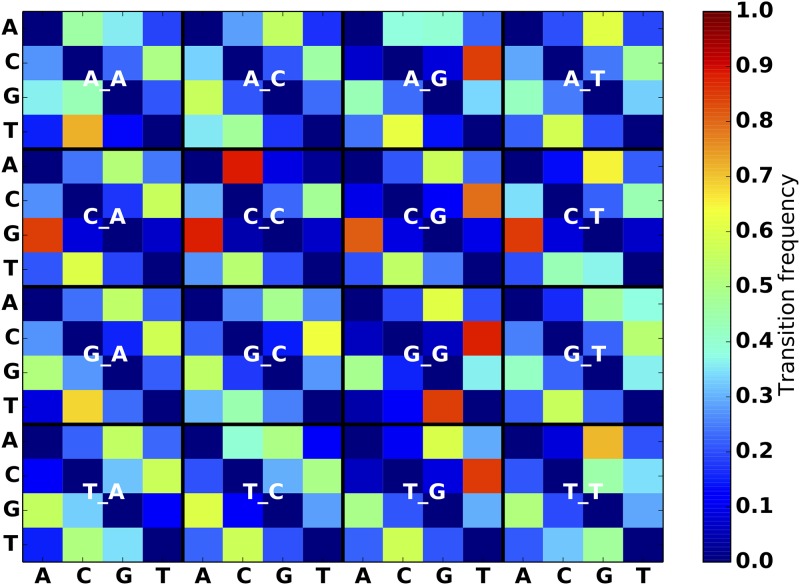
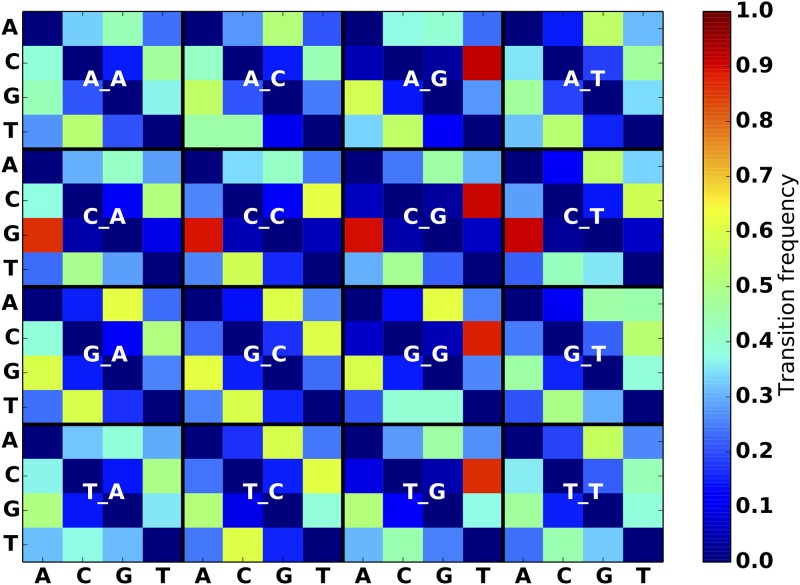
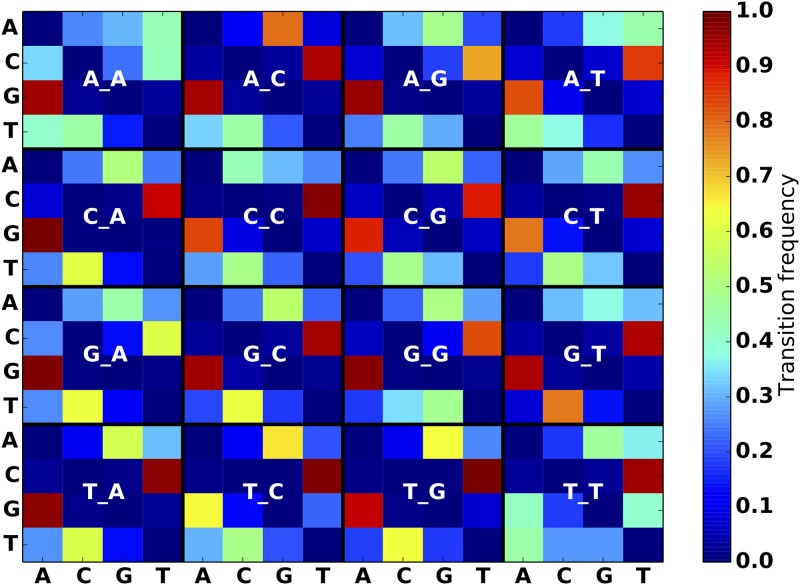
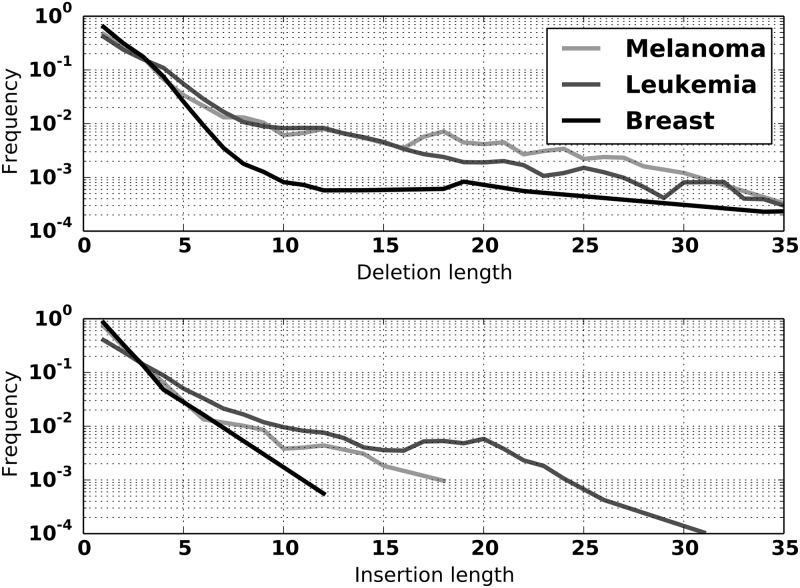
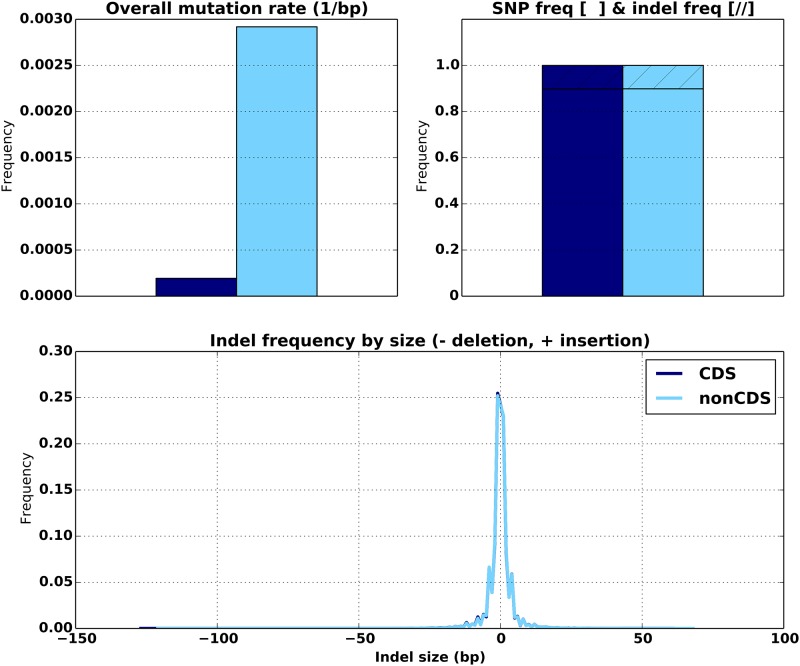
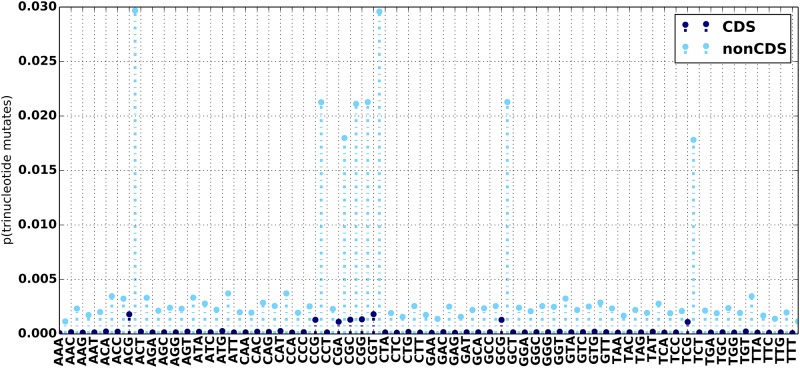
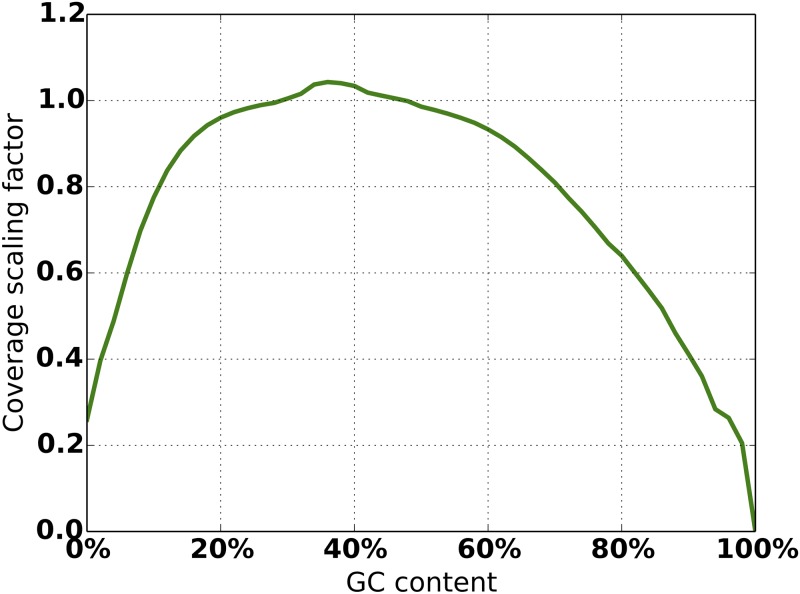
An obstacle to validating and benchmarking methods for genome analysis is that there are few reference datasets available for which the "ground truth" about the mutational landscape of the sample genome is known and fully validated. Additionally, the free and public availability of real human genome datasets is incompatible with the preservation of donor privacy. In order to better analyze and understand genomic data, we need test datasets that model all variants, reflecting known biology as well as sequencing artifacts. Read simulators can fulfill this requirement, but are often criticized for limited resemblance to true data and overall inflexibility. We present NEAT (NExt-generation sequencing Analysis Toolkit), a set of tools that not only includes an easy-to-use read simulator, but also scripts to facilitate variant comparison and tool evaluation. NEAT has a wide variety of tunable parameters which can be set manually on the default model or parameterized using real datasets. The software is freely available at github.com/zstephens/neat-genreads.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
