

Discovery and genotyping of structural variation from long-read haploid genome sequence data
- PMID: 27895111
- PMCID: PMC5411763
- DOI: 10.1101/gr.214007.116
Discovery and genotyping of structural variation from long-read haploid genome sequence data
Erratum in
-
Corrigendum: Discovery and genotyping of structural variation from long-read haploid genome sequence data.Genome Res. 2018 Jan;28(1):144. doi: 10.1101/gr.233007.117. Genome Res. 2018. PMID: 29295848 Free PMC article. No abstract available.
Abstract
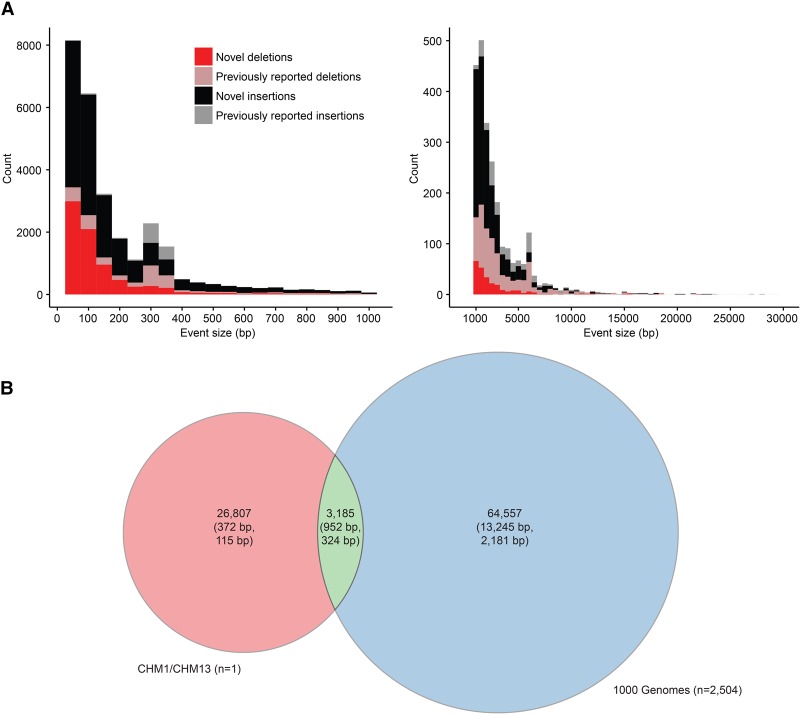
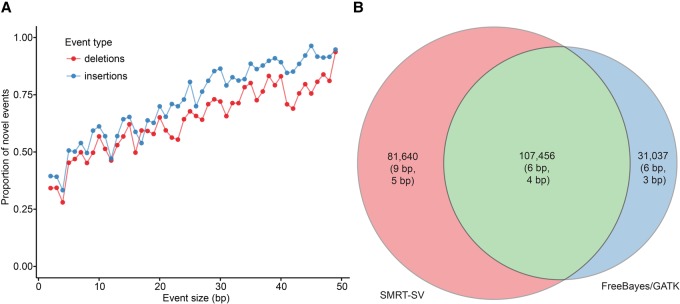
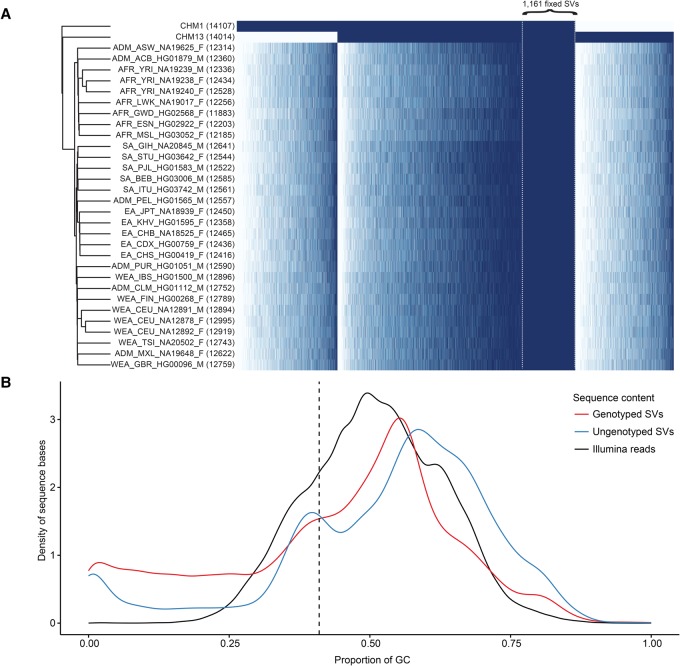
In an effort to more fully understand the full spectrum of human genetic variation, we generated deep single-molecule, real-time (SMRT) sequencing data from two haploid human genomes. By using an assembly-based approach (SMRT-SV), we systematically assessed each genome independently for structural variants (SVs) and indels resolving the sequence structure of 461,553 genetic variants from 2 bp to 28 kbp in length. We find that >89% of these variants have been missed as part of analysis of the 1000 Genomes Project even after adjusting for more common variants (MAF > 1%). We estimate that this theoretical human diploid differs by as much as ∼16 Mbp with respect to the human reference, with long-read sequencing data providing a fivefold increase in sensitivity for genetic variants ranging in size from 7 bp to 1 kbp compared with short-read sequence data. Although a large fraction of genetic variants were not detected by short-read approaches, once the alternate allele is sequence-resolved, we show that 61% of SVs can be genotyped in short-read sequence data sets with high accuracy. Uncoupling discovery from genotyping thus allows for the majority of this missed common variation to be genotyped in the human population. Interestingly, when we repeat SV detection on a pseudodiploid genome constructed in silico by merging the two haploids, we find that ∼59% of the heterozygous SVs are no longer detected by SMRT-SV. These results indicate that haploid resolution of long-read sequencing data will significantly increase sensitivity of SV detection.
© 2017 Huddleston et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Berlin K, Koren S, Chin CS, Drake JP, Landolin JM, Phillippy AM. 2015. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat Biotechnol 33: 623–630. - PubMed
-
- Bhangale TR, Rieder MJ, Livingston RJ, Nickerson DA. 2005. Comprehensive identification and characterization of diallelic insertion-deletion polymorphisms in 330 human candidate genes. Hum Mol Genet 14: 59–69. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous