

Expanding the spectrum of HEXA mutations in Indian patients with Tay-Sachs disease
- PMID: 27896118
- PMCID: PMC5121347
- DOI: 10.1016/j.ymgmr.2014.09.004
Expanding the spectrum of HEXA mutations in Indian patients with Tay-Sachs disease
Abstract
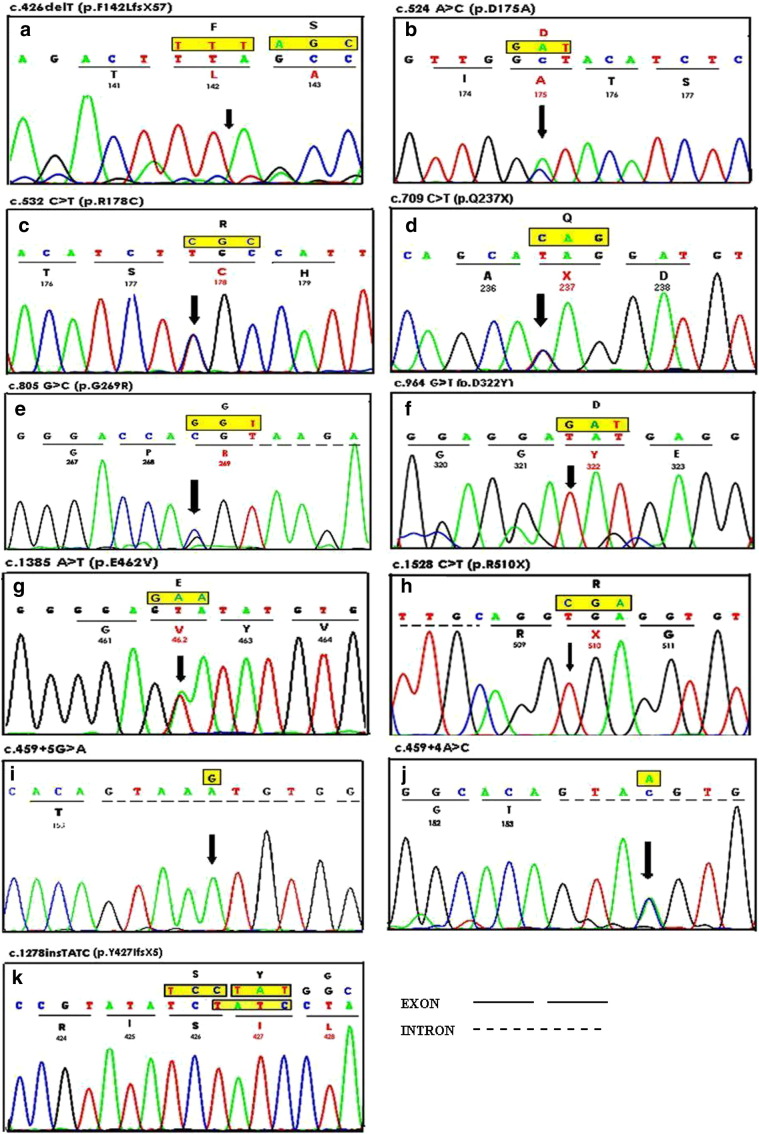
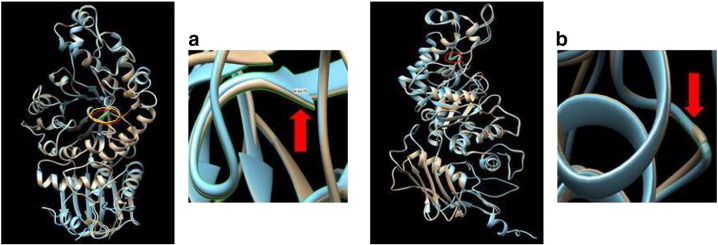
Tay-Sachs disease is an autosomal recessive neurodegenerative disorder occurring due to impaired activity of β-hexosaminidase-A (EC 3.2.1.52), resulting from the mutation in HEXA gene. Very little is known about the molecular pathology of TSD in Indian children except for a few mutations identified by us. The present study is aimed to determine additional mutations leading to Tay-Sachs disease in nine patients confirmed by the deficiency of β-hexosaminidase-A (< 2% of total hexosaminidase activity for infantile patients) in leucocytes. The enzyme activity was assessed by using substrates 4-methylumbelliferyl-N-acetyl-β-d-glucosamine and 4-methylumbelliferyl-N-acetyl-β-d-glucosamine-6-sulfate for total-hexosaminidase and hexosaminidase-A respectively, and heat inactivation method for carrier detection. The exons and exon-intron boundaries of the HEXA gene were bi-directionally sequenced on an automated sequencer. 'In silico' analyses for novel mutations were carried out using SIFT, Polyphen2 and MutationT@ster software programs. The structural study was carried out by UCSF Chimera software using the crystallographic structure of β-hexosaminidase-A (PDB-ID: 2GJX) as the template. Our study identified four novel mutations in three cases. These include a compound heterozygous missense mutation c.524A>C (D175A) and c.805G>C (p.G269R) in one case; and one small 1 bp deletion c.426delT (p.F142LfsX57) and one splice site mutation c.459+4A>C in the other two cases respectively. None of these mutations were detected in 100 chromosomes from healthy individuals of the same ethnic group. Three previously reported missense mutations, (i) c.532C>T (p.R178C), (ii) c.964G>T (p.D322Y), and (iii) c.1385A>T (p.E462V); two nonsense mutations (i) c.709C>T (p.Q237X) and (ii) c.1528C>T (p.R510X), one 4 bp insertion c.1277_1278insTATC (p.Y427IfsX5) and one splice site mutation c.459+5G>A were also identified in six cases. We observe from this study that novel mutations are more frequently observed in Indian patients with Tay-Sachs disease with clustering of ~ 73% of disease causing mutations in exons 5 to 12. This database can be used for a carrier rate screening in the larger population of the country.
Keywords: HEXA gene; Hex-A, β-Hexosaminidase A; LSDs, Lysosomal storage disorders; Lysosomal enzyme; MUG, 4-Methylumbelliferyl-N-acetyl-β-d-glucosamine; MUGS, 4-Methylumbelliferyl-N-acetyl-β-d-glucosamine-6-sulfate; SD, Sandhoff disease; TSD, Tay–Sachs disease; Tay–Sachs disease; β-Hexosaminidase-A.
Figures
References
-
- Sheth J., Mistri M., Sheth F., Shah R., Bavdekar A., Godbole K., Nanavaty N., Datar C., Kamate M., Oza N., Ankleshwaria C., Mehta S., Jackson M. JIMD Rep. 2013. Burden of lysosomal storage disorders in India: experience of 387 affected children from a single diagnostic facility. (Epub ahead of print) - PMC - PubMed
-
- Tanaka A., Fujimaru M., Choeh K., Isshiki G. Novel mutations, including the second most common in Japan, in the B-hexosaminidase A subunit gene, a simple screening of Japanese patients with Tay–Sachs disease. J. Hum. Genet. 1999;44:91–95. - PubMed
-
- Fernades Filho J.A., Shapiro B.E. Tay–Sachs disease. Arch. Neurol. 2004;61:1466–1468. - PubMed
-
- Myerowitz R., Costigan F.C. The major defect in Ashkenazi Jews with Tay–Sachs disease is an insertion in the gene for alpha-chain of beta-hexosaminidase. J. Biol. Chem. 1988;263:18587–18589. - PubMed
-
- Kaback M.M., Lim-steele J.S., Dabholkar D., Brown D., Levy N., Zeiger K. Tay–Sachs disease carrier screening, prenatal diagnosis and the molecular era. An international perspective, 1970–1993. The International TSD data collection Network. JAMA. 1993;270:2307–2315. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
