

Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer
- PMID: 27897170
- PMCID: PMC5141345
- DOI: 10.1038/ncomms13668
Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer
Abstract
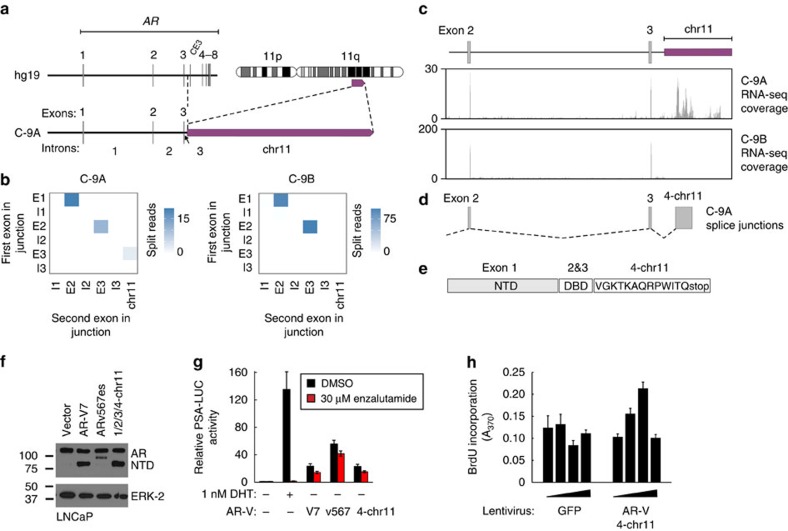
Molecularly targeted therapies for advanced prostate cancer include castration modalities that suppress ligand-dependent transcriptional activity of the androgen receptor (AR). However, persistent AR signalling undermines therapeutic efficacy and promotes progression to lethal castration-resistant prostate cancer (CRPC), even when patients are treated with potent second-generation AR-targeted therapies abiraterone and enzalutamide. Here we define diverse AR genomic structural rearrangements (AR-GSRs) as a class of molecular alterations occurring in one third of CRPC-stage tumours. AR-GSRs occur in the context of copy-neutral and amplified AR and display heterogeneity in breakpoint location, rearrangement class and sub-clonal enrichment in tumours within and between patients. Despite this heterogeneity, one common outcome in tumours with high sub-clonal enrichment of AR-GSRs is outlier expression of diverse AR variant species lacking the ligand-binding domain and possessing ligand-independent transcriptional activity. Collectively, these findings reveal AR-GSRs as important drivers of persistent AR signalling in CRPC.
Conflict of interest statement
S.R.P. and S.M.D. have served as a paid consultant/advisor for Medivation/Astellas. The remaining authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
