

High-Resolution Phenotypic Landscape of the RNA Polymerase II Trigger Loop
- PMID: 27898685
- PMCID: PMC5127505
- DOI: 10.1371/journal.pgen.1006321
High-Resolution Phenotypic Landscape of the RNA Polymerase II Trigger Loop
Erratum in
-
Correction: High-Resolution Phenotypic Landscape of the RNA Polymerase II Trigger Loop.PLoS Genet. 2018 Jan 3;14(1):e1007158. doi: 10.1371/journal.pgen.1007158. eCollection 2018 Jan. PLoS Genet. 2018. PMID: 29298339 Free PMC article.
Abstract
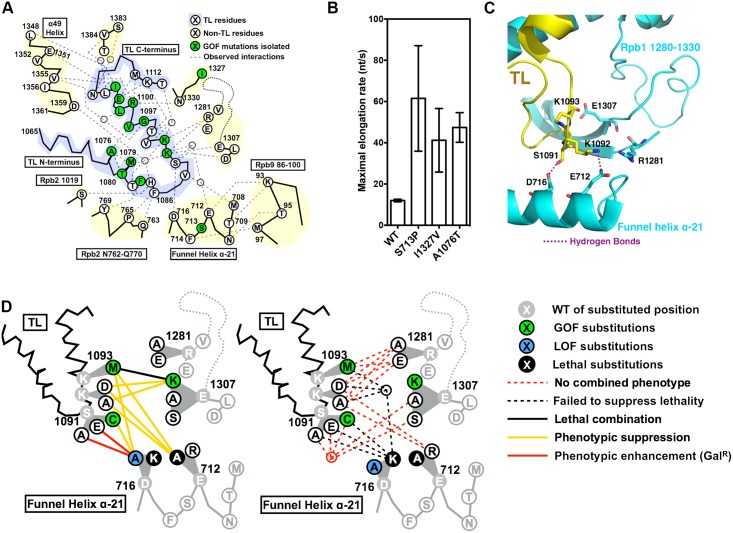
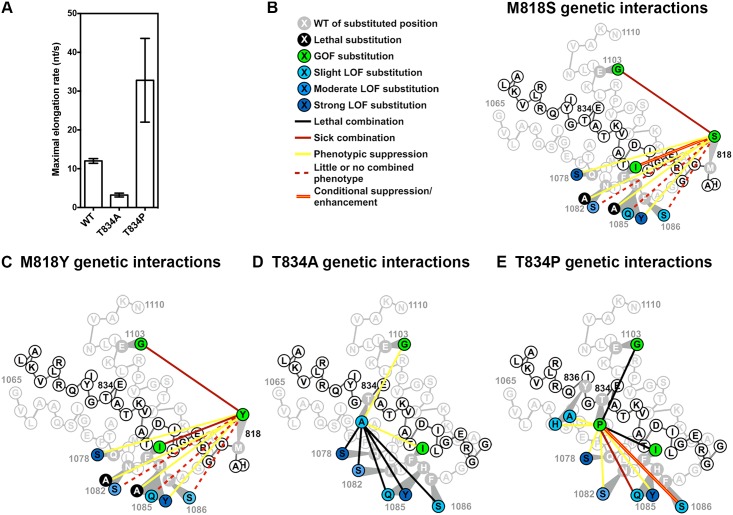
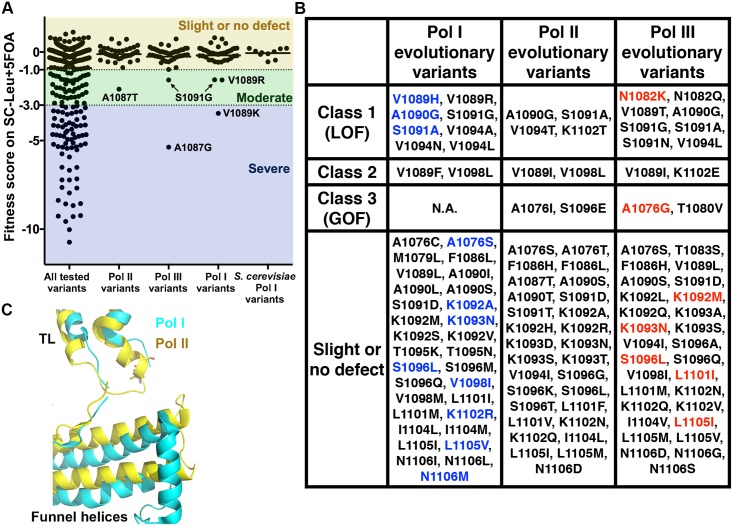
The active sites of multisubunit RNA polymerases have a "trigger loop" (TL) that multitasks in substrate selection, catalysis, and translocation. To dissect the Saccharomyces cerevisiae RNA polymerase II TL at individual-residue resolution, we quantitatively phenotyped nearly all TL single variants en masse. Three mutant classes, revealed by phenotypes linked to transcription defects or various stresses, have distinct distributions among TL residues. We find that mutations disrupting an intra-TL hydrophobic pocket, proposed to provide a mechanism for substrate-triggered TL folding through destabilization of a catalytically inactive TL state, confer phenotypes consistent with pocket disruption and increased catalysis. Furthermore, allele-specific genetic interactions among TL and TL-proximal domain residues support the contribution of the funnel and bridge helices (BH) to TL dynamics. Our structural genetics approach incorporates structural and phenotypic data for high-resolution dissection of transcription mechanisms and their evolution, and is readily applicable to other essential yeast proteins.
Conflict of interest statement
Authors JVdB and RS are employed by MorphoSys AG, which currently holds rights to the Slonomics DNA synthesis technology utilized in the manuscript.
Figures
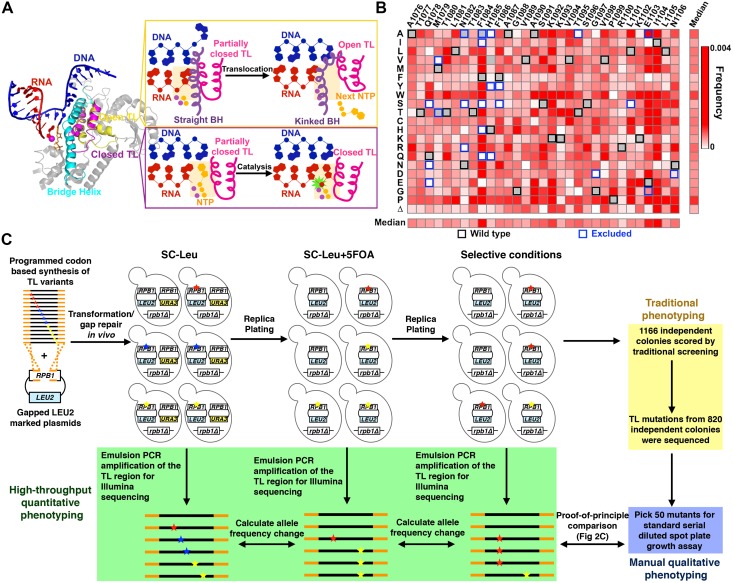
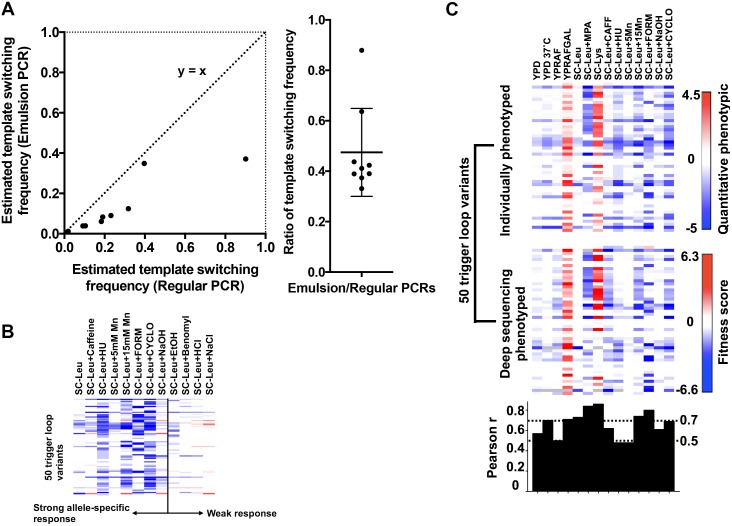
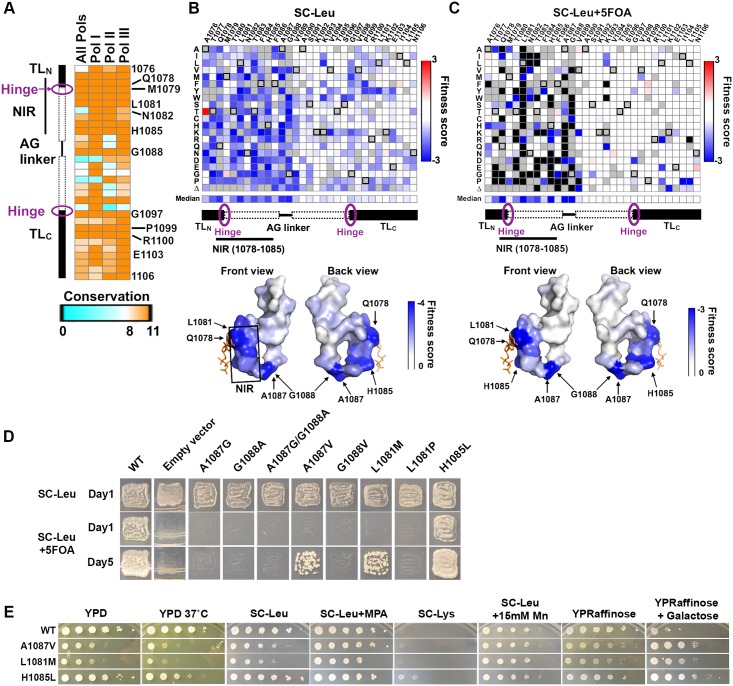
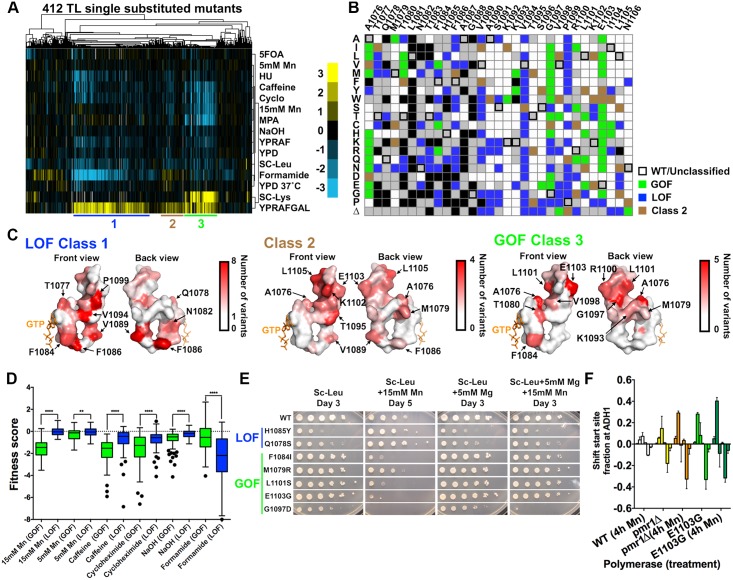
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
