

Genetic predisposition to kidney cancer
- PMID: 27899189
- PMCID: PMC5137802
- DOI: 10.1053/j.seminoncol.2016.09.001
Genetic predisposition to kidney cancer
Abstract
Kidney cancer is not a single disease but is made up of a number of different types of cancer classified by histology that are disparate in presentation, clinical course, and genetic basis. Studies of families with inherited renal cell carcinoma (RCC) have provided the basis for our understanding of the causative genes and altered metabolic pathways in renal cancer with different histologies. Von Hippel-Lindau disease was the first renal cancer disorder with a defined genetic basis. Over the next two decades, the genes responsible for a number of other inherited renal cancer syndromes including hereditary papillary renal carcinoma, Birt-Hogg-Dube´syndrome, hereditary leiomyomatosis and renal cell carcinoma, and succinate dehydrogenase-associated renal cancer were identified. Recently, renal cell carcinoma has been confirmed as part of the clinical phenotype in individuals from families with BAP1-associated tumor predisposition syndrome and MiTF-associated cancer syndrome. Here we summarize the clinical characteristics of and causative genes for these and other inherited RCC syndromes, the pathways that are dysregulated when the inherited genes are mutated, and recommended clinical management of patients with these inherited renal cancer syndromes.
Keywords: Birt-Hogg-Dubé syndrome; Hereditary leiomyomatosis and renal cell carcinoma; Hereditary papillary renal carcinoma; Inherited renal cancer syndromes; Kidney neoplasms; Von Hippel-Lindau.
Published by Elsevier Inc.
Conflict of interest statement
None.
Figures

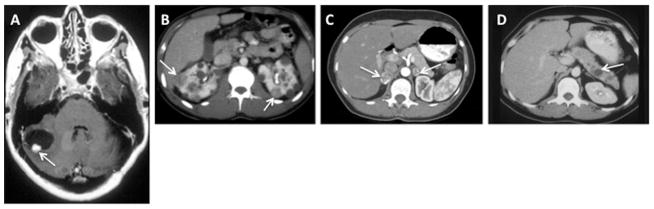
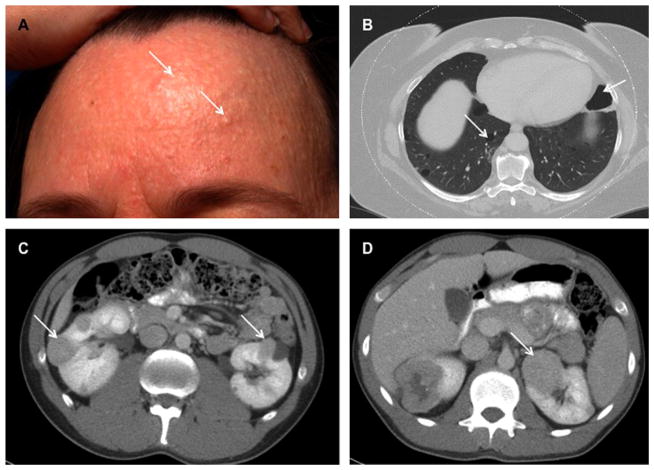
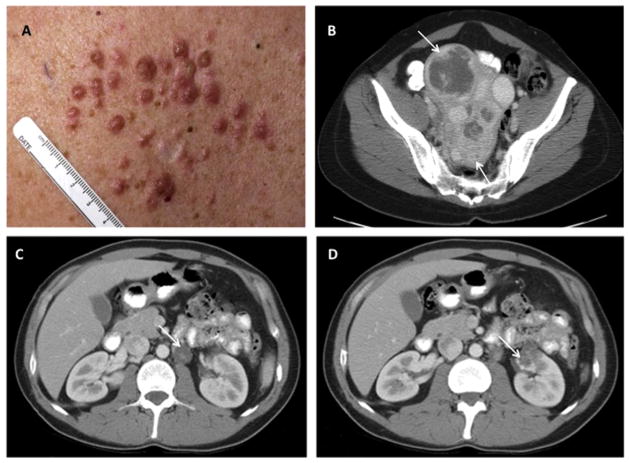
References
-
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. - PubMed
-
- Nordstrom-O’Brien M, van der Luijt RB, van Rooijen E, et al. Genetic analysis of von Hippel-Lindau disease. Hum Mutat. 2010;31:521–37. - PubMed
-
- Walther MM, Lubensky IA, Venzon D, Zbar B, Linehan WM. Prevalence of microscopic lesions in grossly normal renal parenchyma from patients with von Hippel-Lindau disease, sporadic renal cell carcinoma and no renal disease: clinical implications. J Urol. 1995;154:2010–4. - PubMed
-
- Walther MM, Choyke PL, Glenn G, et al. Renal cancer in families with hereditary renal cancer: prospective analysis of a tumor size threshold for renal parenchymal sparing surgery. J Urol. 1999;161:1475–9. - PubMed
-
- Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–20. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
