

Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria
- PMID: 27899527
- PMCID: PMC5231411
- DOI: 10.1126/scisignal.aai8441
Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria
Abstract
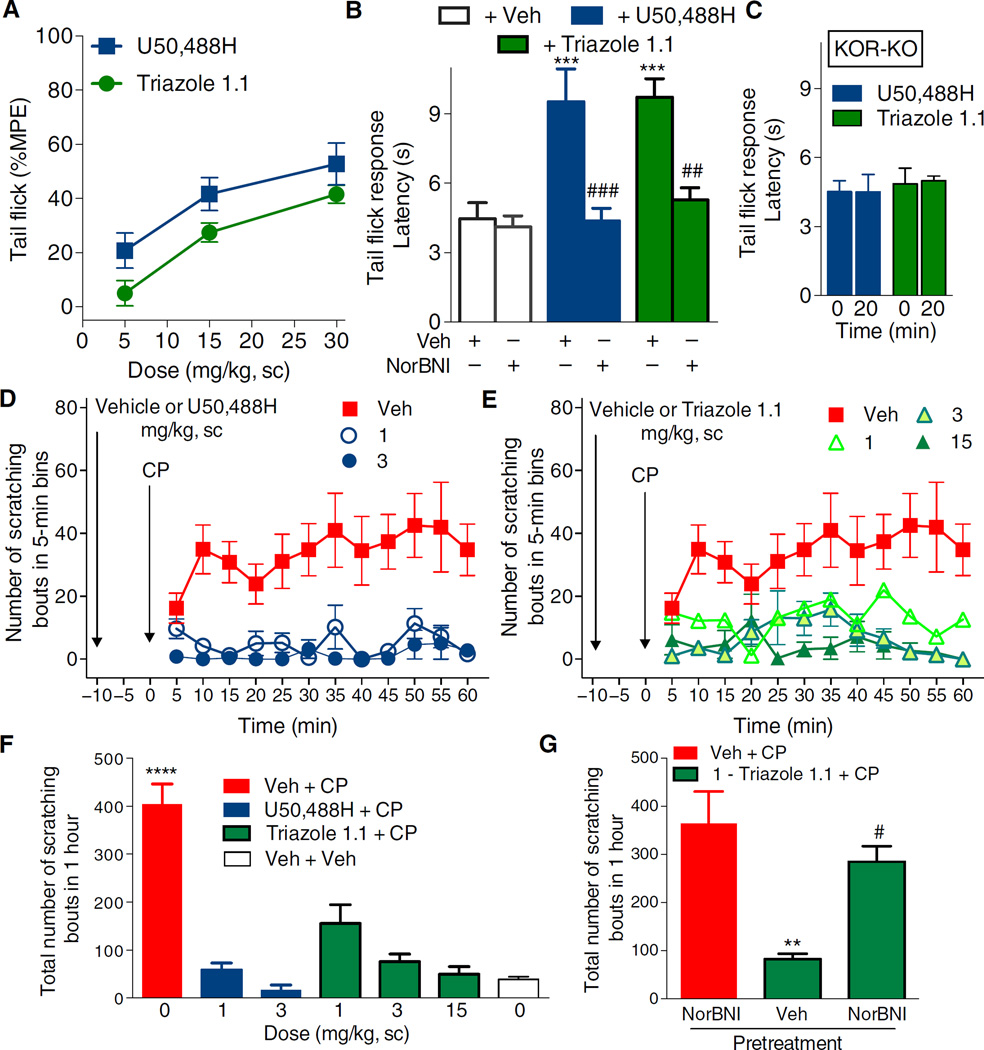
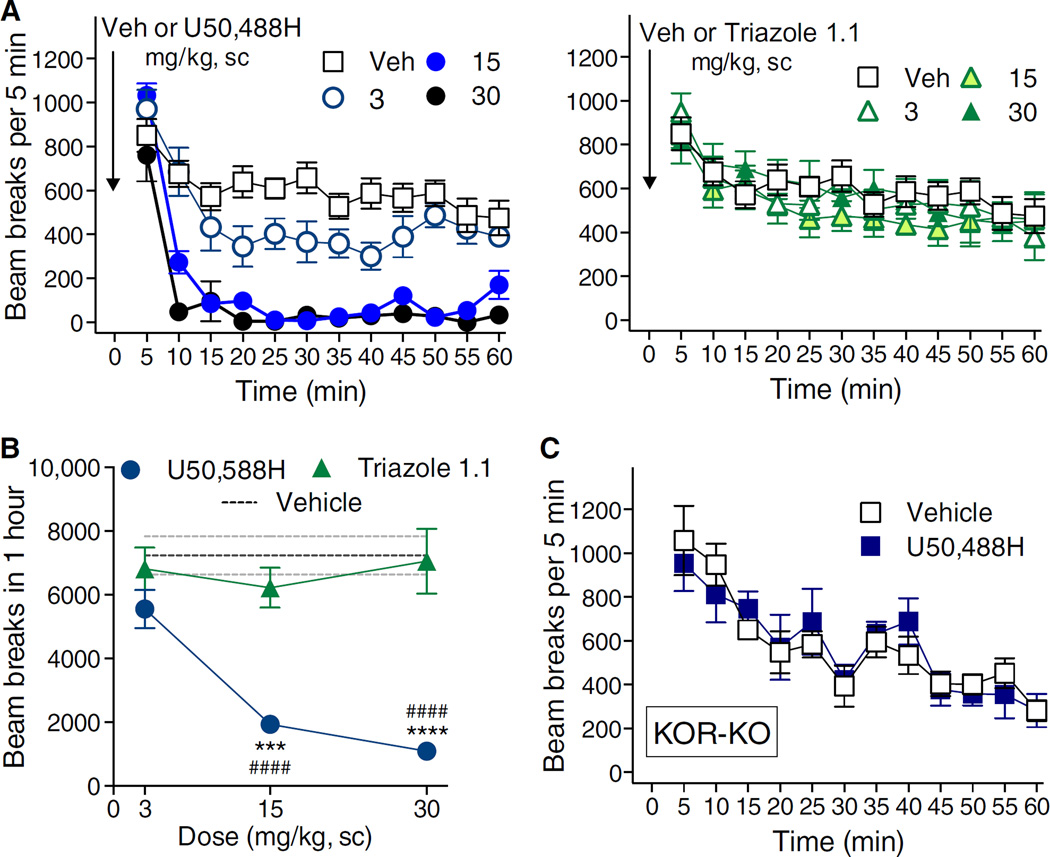
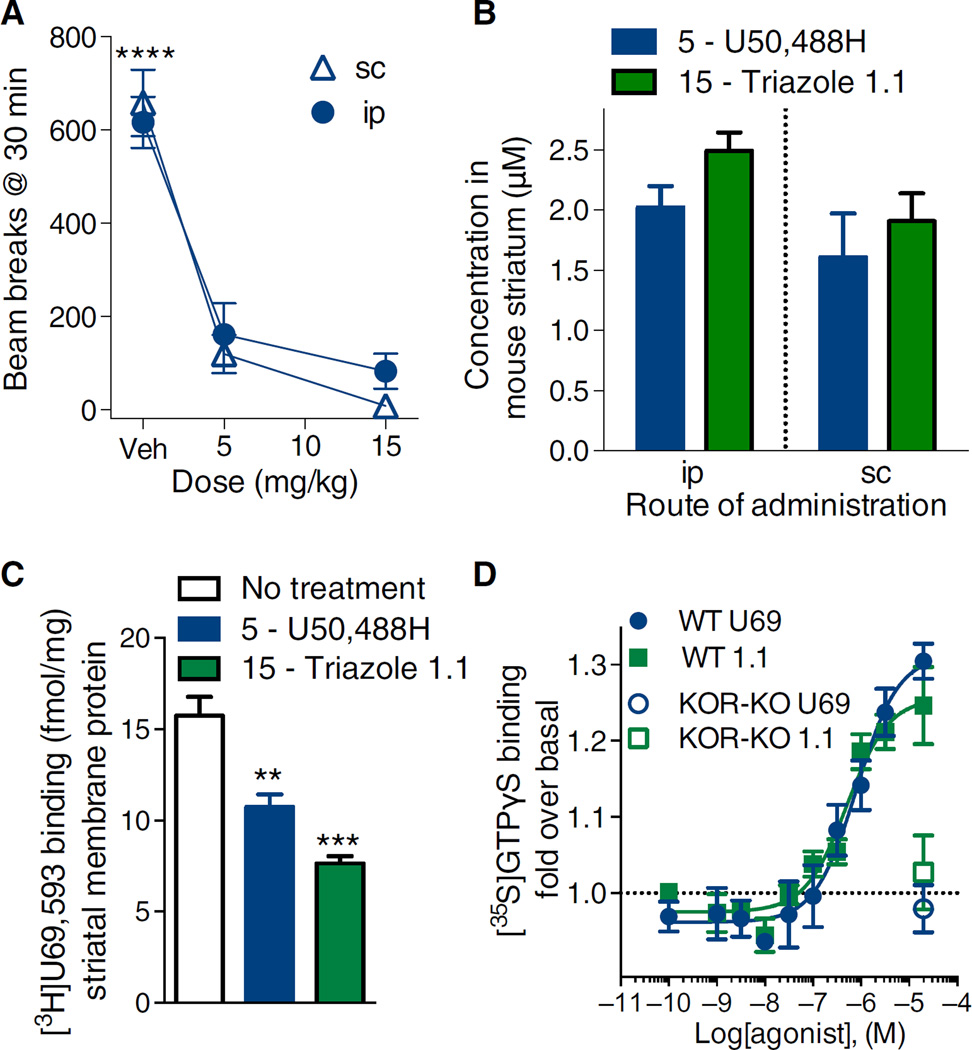
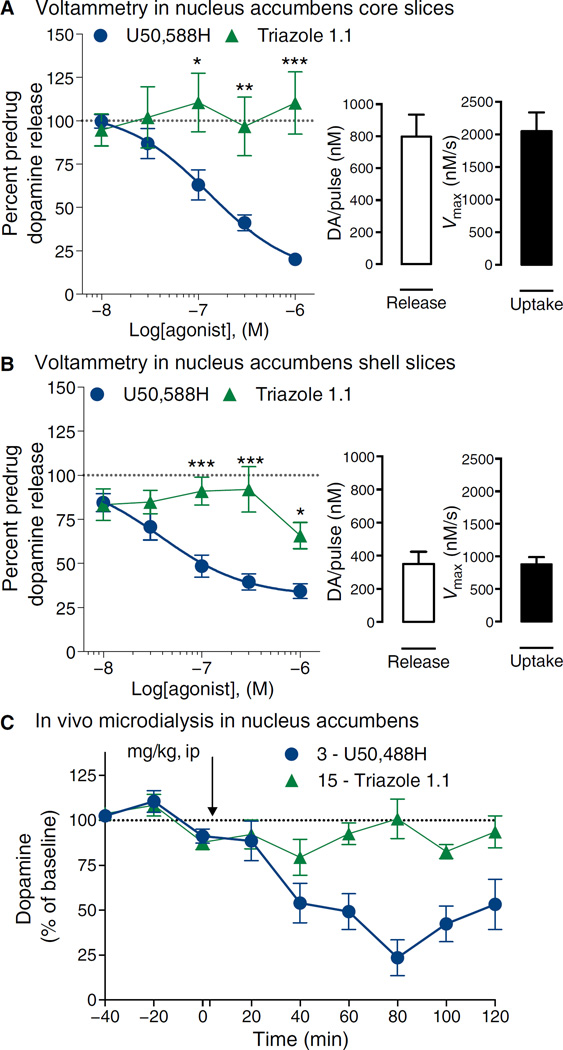
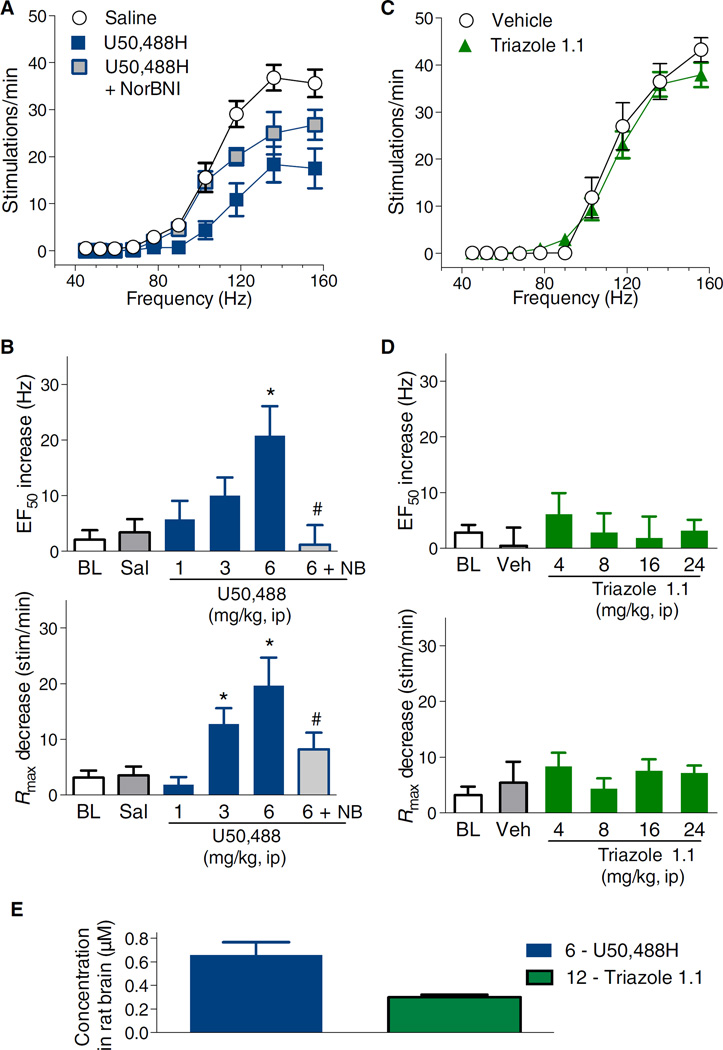
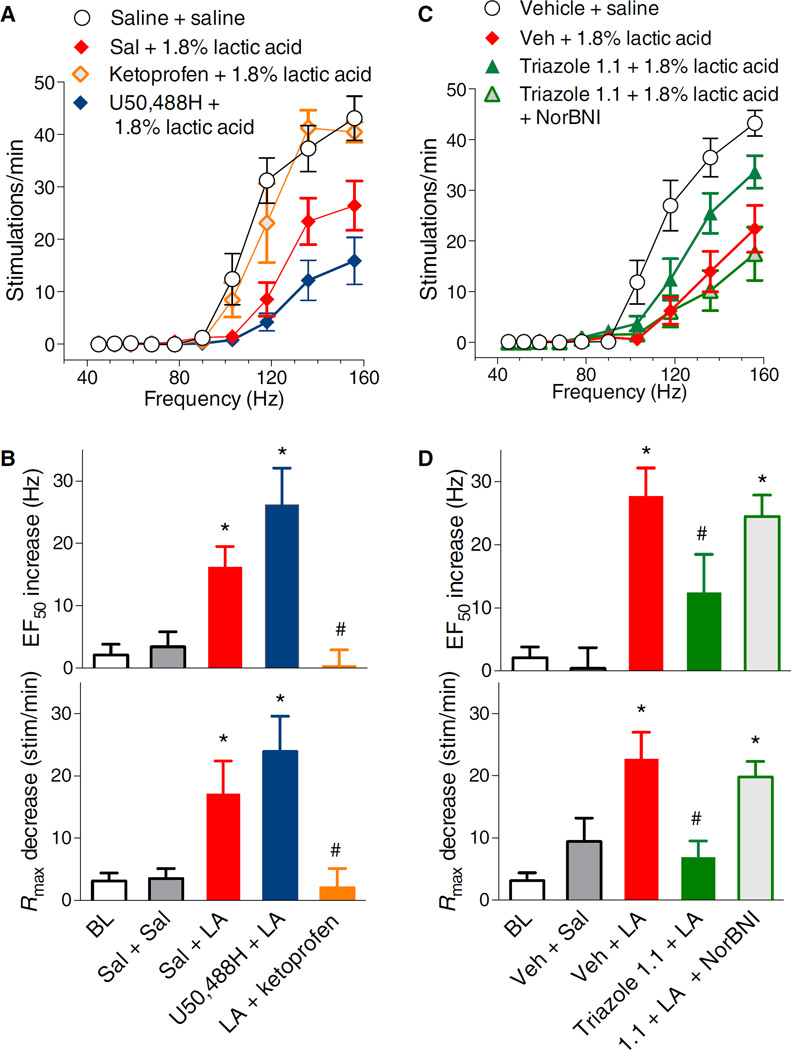
Agonists targeting the kappa opioid receptor (KOR) have been promising therapeutic candidates because of their efficacy for treating intractable itch and relieving pain. Unlike typical opioid narcotics, KOR agonists do not produce euphoria or lead to respiratory suppression or overdose. However, they do produce dysphoria and sedation, side effects that have precluded their clinical development as therapeutics. KOR signaling can be fine-tuned to preferentially activate certain pathways over others, such that agonists can bias signaling so that the receptor signals through G proteins rather than other effectors such as βarrestin2. We evaluated a newly developed G protein signaling-biased KOR agonist in preclinical models of pain, pruritis, sedation, dopamine regulation, and dysphoria. We found that triazole 1.1 retained the antinociceptive and antipruritic efficacies of a conventional KOR agonist, yet it did not induce sedation or reductions in dopamine release in mice, nor did it produce dysphoria as determined by intracranial self-stimulation in rats. These data demonstrated that biased agonists may be used to segregate physiological responses downstream of the receptor. Moreover, the findings suggest that biased KOR agonists may present a means to treat pain and intractable itch without the side effects of dysphoria and sedation and with reduced abuse potential.
Copyright © 2016, American Association for the Advancement of Science.
Figures
Comment in
-
New connections: Getting the good without the bad, opiate edition.Sci Signal. 2017 Mar 21;10(471):eaan2516. doi: 10.1126/scisignal.aan2516. Sci Signal. 2017. PMID: 28325819
References
-
- Zhou L, Stahl EL, Lovell KM, Frankowski KJ, Prisinzano TE, Aubé J, Bohn LM. Characterization of kappa opioid receptor mediated, dynorphin-stimulated [35S]GTPγS binding in mouse striatum for the evaluation of selective KOR ligands in an endogenous setting. Neuropharmacology. 2015;99:131–141. - PMC - PubMed
-
- Chavkin C, James IF, Goldstein A. Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science. 1982;215:413–415. - PubMed
-
- Mansour A, Khachaturian H, Lewis ME, Akil H, Watson SJ. Anatomy of CNS opioid receptors. Trends Neurosci. 1988;11:308–314. - PubMed
-
- Kivell B, Prisinzano TE. Kappa opioids and the modulation of pain. Psychopharmacology (Berl) 2010;210:109–119. - PubMed
-
- Cowan A, Kehner GB, Inan S. Targeting itch with ligands selective for κ opioid receptors. Handb. Exp. Pharmacol. 2015;226:291–314. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
