

Alignment-free $d_2^*$ oligonucleotide frequency dissimilarity measure improves prediction of hosts from metagenomically-derived viral sequences
- PMID: 27899557
- PMCID: PMC5224470
- DOI: 10.1093/nar/gkw1002
Alignment-free $d_2^*$ oligonucleotide frequency dissimilarity measure improves prediction of hosts from metagenomically-derived viral sequences
Abstract
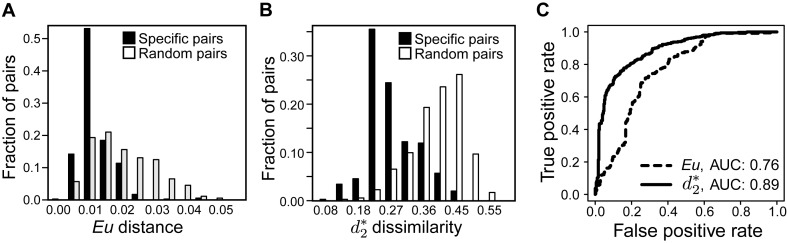
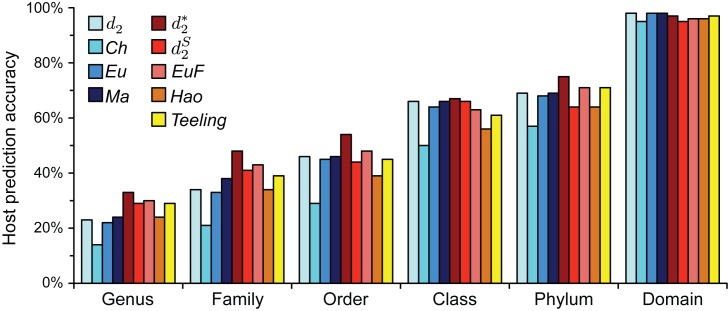
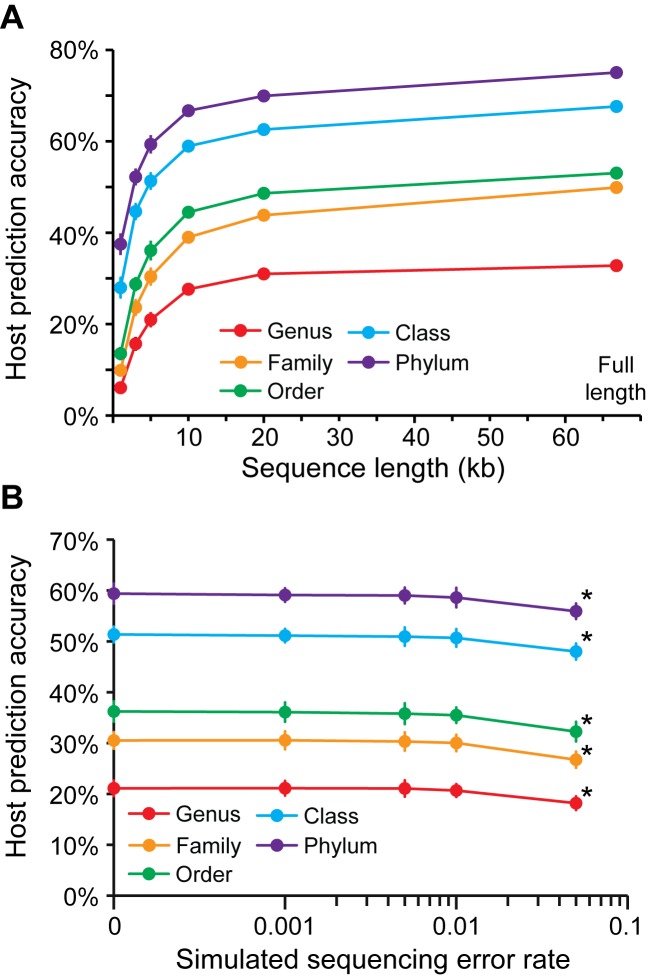
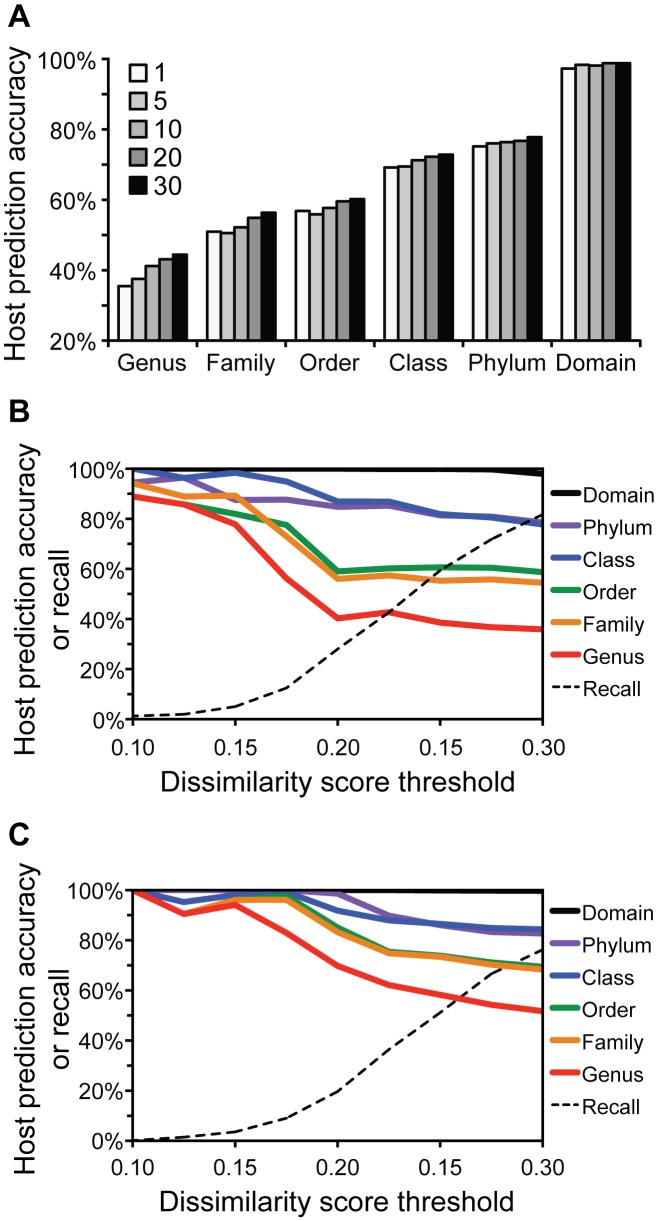
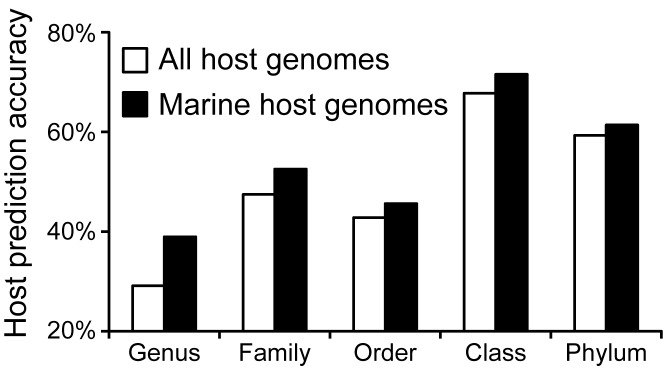
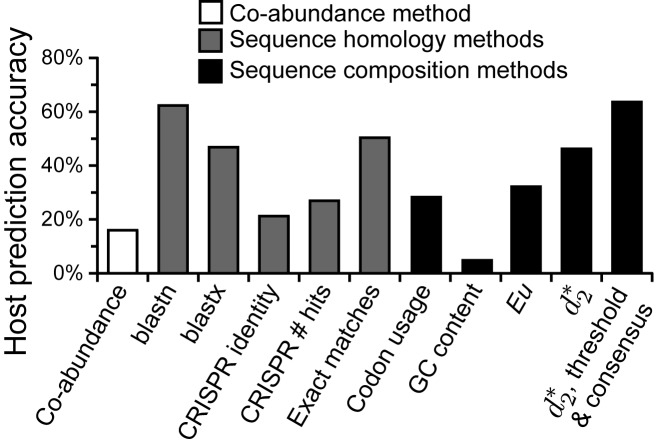
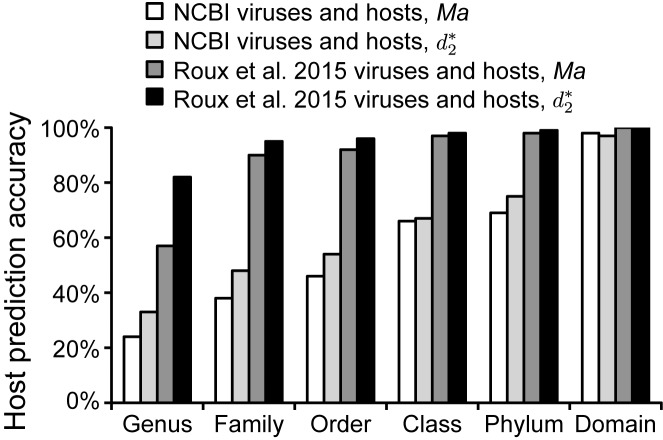
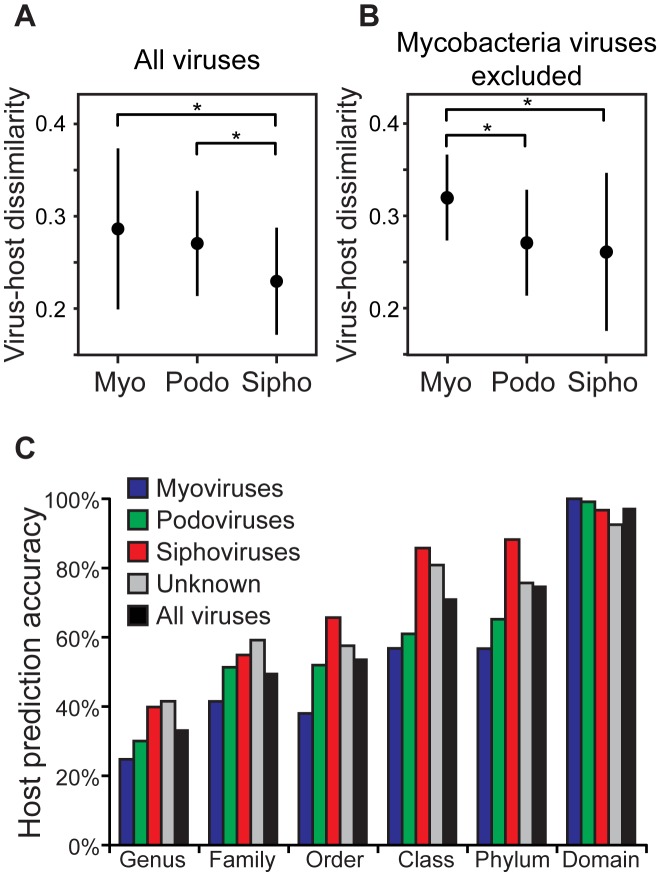
Viruses and their host genomes often share similar oligonucleotide frequency (ONF) patterns, which can be used to predict the host of a given virus by finding the host with the greatest ONF similarity. We comprehensively compared 11 ONF metrics using several k-mer lengths for predicting host taxonomy from among ∼32 000 prokaryotic genomes for 1427 virus isolate genomes whose true hosts are known. The background-subtracting measure [Formula: see text] at k = 6 gave the highest host prediction accuracy (33%, genus level) with reasonable computational times. Requiring a maximum dissimilarity score for making predictions (thresholding) and taking the consensus of the 30 most similar hosts further improved accuracy. Using a previous dataset of 820 bacteriophage and 2699 bacterial genomes, [Formula: see text] host prediction accuracies with thresholding and consensus methods (genus-level: 64%) exceeded previous Euclidian distance ONF (32%) or homology-based (22-62%) methods. When applied to metagenomically-assembled marine SUP05 viruses and the human gut virus crAssphage, [Formula: see text]-based predictions overlapped (i.e. some same, some different) with the previously inferred hosts of these viruses. The extent of overlap improved when only using host genomes or metagenomic contigs from the same habitat or samples as the query viruses. The [Formula: see text] ONF method will greatly improve the characterization of novel, metagenomic viruses.
© The Author(s) 2016. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Rappé M.S., Giovannoni S.J. The uncultured microbial majority. Annu. Rev. Microbiol. 2003;57:369–394. - PubMed
-
- Breitbart M., Rohwer F. Here a virus, there a virus, everywhere the same virus. Trends Microbiol. 2005;13:278–284. - PubMed
-
- Fuhrman J.A. Marine viruses and their biogeochemical and ecological effects. Nature. 1999;399:541–548. - PubMed
-
- Weinbauer M.G. Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 2004;28:127–181. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
