

Bainbridge-Ropers syndrome caused by loss-of-function variants in ASXL3: a recognizable condition
- PMID: 27901041
- PMCID: PMC5255962
- DOI: 10.1038/ejhg.2016.165
Bainbridge-Ropers syndrome caused by loss-of-function variants in ASXL3: a recognizable condition
Abstract
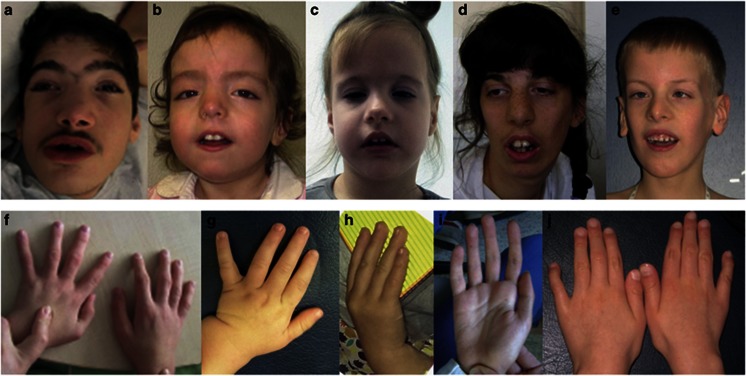
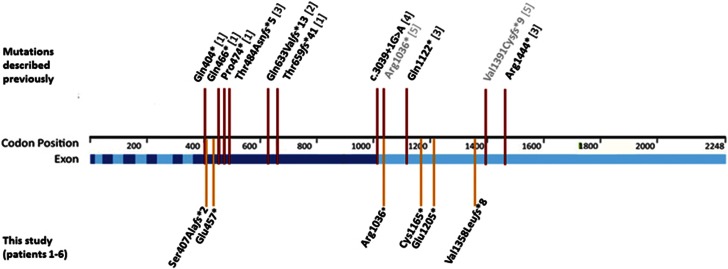
Truncating ASXL3 mutations were first identified in 2013 by Bainbridge et al. as a cause of syndromic intellectual disability in four children with similar phenotypes using whole-exome sequencing. The clinical features - postulated by Bainbridge et al. to be overlapping with Bohring-Opitz syndrome - were developmental delay, severe feeding difficulties, failure to thrive and neurological abnormalities. This condition was included in OMIM as 'Bainbridge-Ropers syndrome' (BRPS, #615485). To date, a total of nine individuals with BRPS have been published in the literature in four reports (Bainbridge et al., Dinwiddie et al, Srivastava et al. and Hori et al.). In this report, we describe six unrelated patients with newly diagnosed heterozygous de novo loss-of-function variants in ASXL3 and concordant clinical features: severe muscular hypotonia with feeding difficulties in infancy, significant motor delay, profound speech impairment, intellectual disability and a characteristic craniofacial phenotype (long face, arched eyebrows with mild synophrys, downslanting palpebral fissures, prominent columella, small alae nasi, high, narrow palate and relatively little facial expression). The majority of key features characteristic for Bohring-Opitz syndrome were absent in our patients (eg, the typical posture of arms, intrauterine growth retardation, microcephaly, trigonocephaly, typical facial gestalt with nevus flammeus of the forehead and exophthalmos). Therefore we emphasize that BRPS syndrome, caused by ASXL3 loss-of-function variants, is a clinically distinct intellectual disability syndrome with a recognizable phenotype distinguishable from that of Bohring-Opitz syndrome.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
