

Adenosine Kinase Deficiency in the Brain Results in Maladaptive Synaptic Plasticity
- PMID: 27903722
- PMCID: PMC5148215
- DOI: 10.1523/JNEUROSCI.2146-16.2016
Adenosine Kinase Deficiency in the Brain Results in Maladaptive Synaptic Plasticity
Abstract
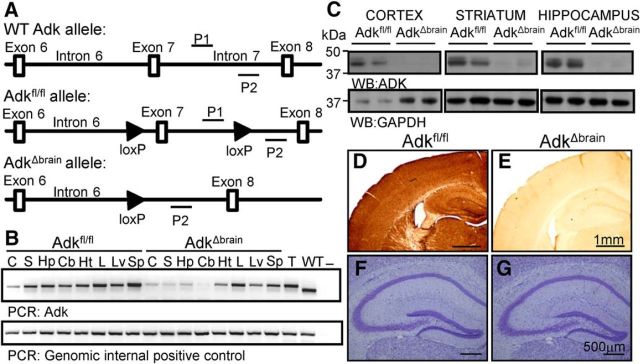
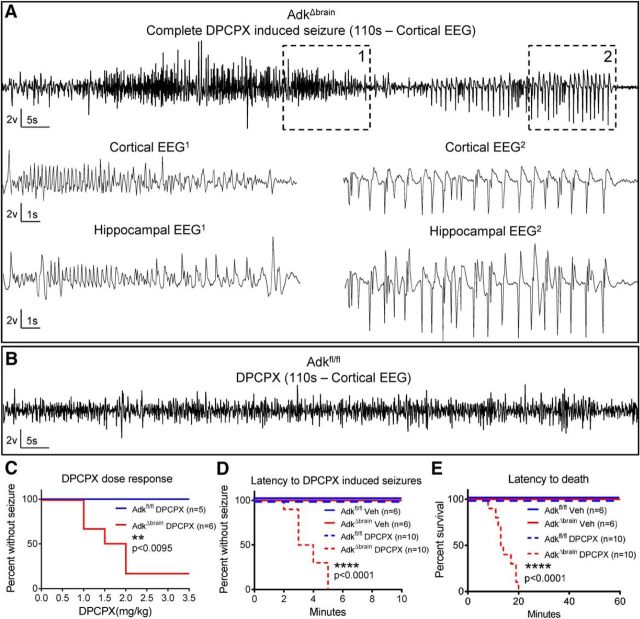
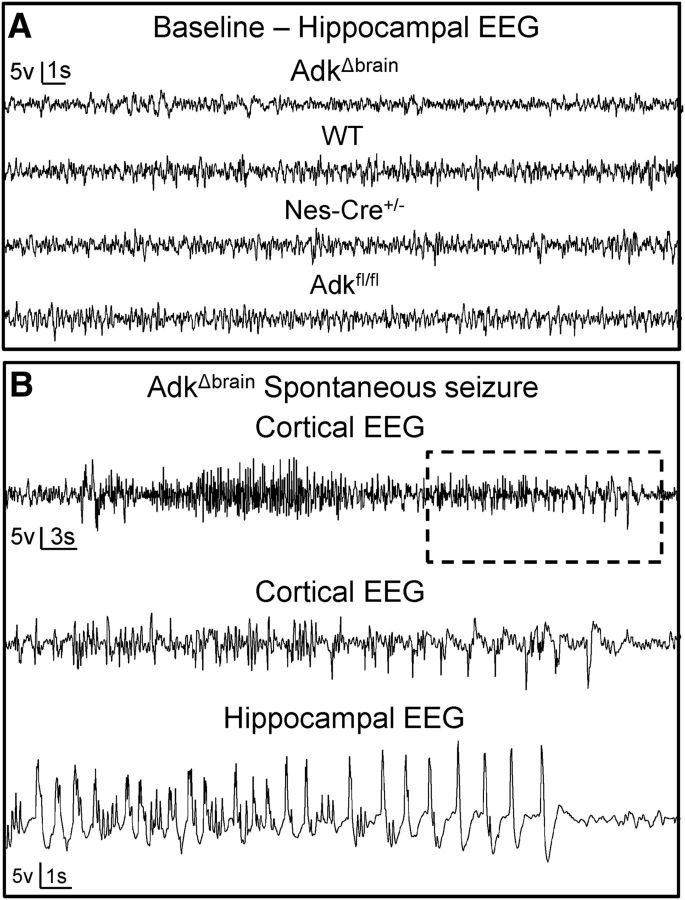
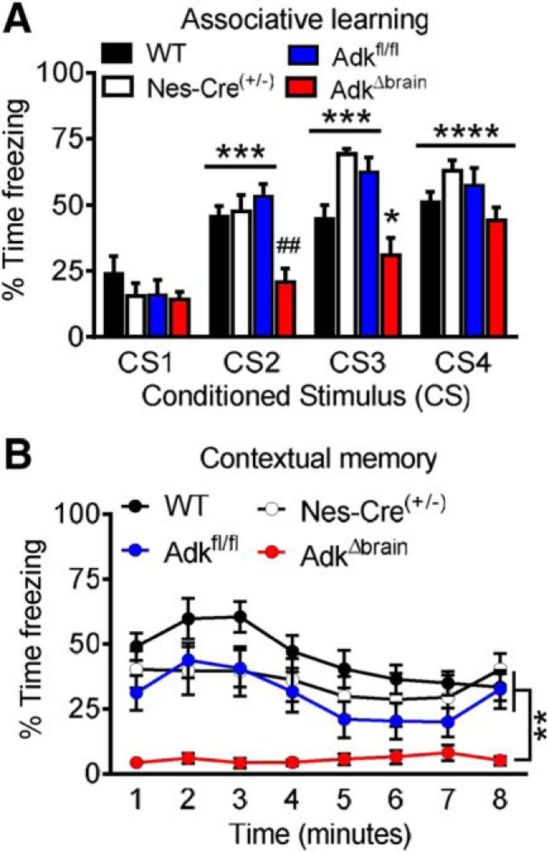
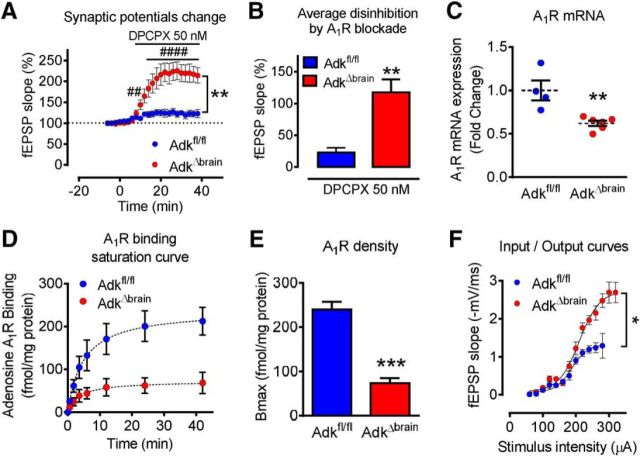
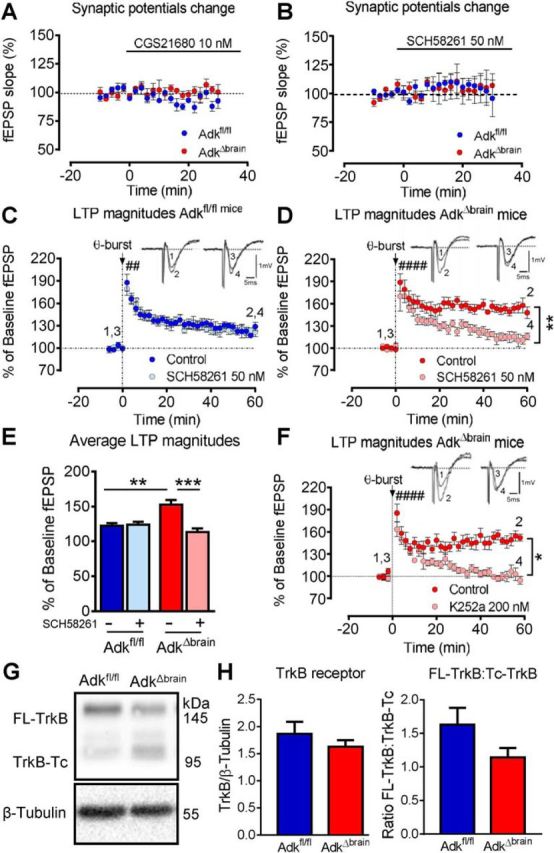
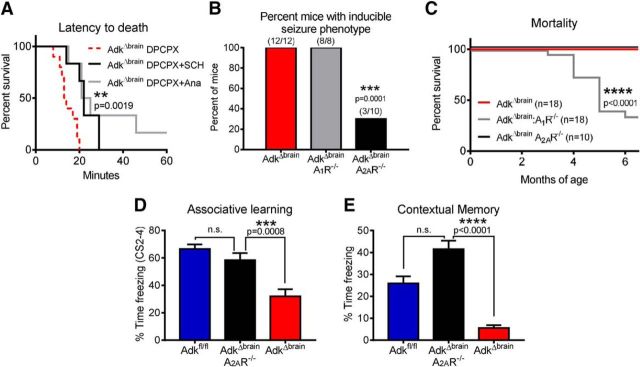
Adenosine kinase (ADK) deficiency in human patients (OMIM:614300) disrupts the methionine cycle and triggers hypermethioninemia, hepatic encephalopathy, cognitive impairment, and seizures. To identify whether this neurological phenotype is intrinsically based on ADK deficiency in the brain or if it is secondary to liver dysfunction, we generated a mouse model with a brain-wide deletion of ADK by introducing a Nestin-Cre transgene into a line of conditional ADK deficient Adkfl/fl mice. These AdkΔbrain mice developed a progressive stress-induced seizure phenotype associated with spontaneous convulsive seizures and profound deficits in hippocampus-dependent learning and memory. Pharmacological, biochemical, and electrophysiological studies suggest enhanced adenosine levels around synapses resulting in an enhanced adenosine A1 receptor (A1R)-dependent protective tone despite lower expression levels of the receptor. Theta-burst-induced LTP was enhanced in the mutants and this was dependent on adenosine A2A receptor (A2AR) and tropomyosin-related kinase B signaling, suggesting increased activation of these receptors in synaptic plasticity phenomena. Accordingly, reducing adenosine A2A receptor activity in AdkΔbrain mice restored normal associative learning and contextual memory and attenuated seizure risk. We conclude that ADK deficiency in the brain triggers neuronal adaptation processes that lead to dysregulated synaptic plasticity, cognitive deficits, and increased seizure risk. Therefore, ADK mutations have an intrinsic effect on brain physiology and may present a genetic risk factor for the development of seizures and learning impairments. Furthermore, our data show that blocking A2AR activity therapeutically can attenuate neurological symptoms in ADK deficiency.
Significance statement: A novel human genetic condition (OMIM #614300) that is based on mutations in the adenosine kinase (Adk) gene has been discovered recently. Affected patients develop hepatic encephalopathy, seizures, and severe cognitive impairment. To model and understand the neurological phenotype of the human mutation, we generated a new conditional knock-out mouse with a brain-specific deletion of Adk (AdkΔbrain). Similar to ADK-deficient patients, AdkΔbrain mice develop seizures and cognitive deficits. We identified increased basal synaptic transmission and enhanced adenosine A2A receptor (A2AR)-dependent synaptic plasticity as the underlying mechanisms that govern these phenotypes. Our data show that neurological phenotypes in ADK-deficient patients are intrinsic to ADK deficiency in the brain and that blocking A2AR activity therapeutically can attenuate neurological symptoms in ADK deficiency.
Keywords: adenosine kinase; epilepsy; gene mutation; human genetic disorder; learning and memory; mouse model.
Copyright © 2016 the authors 0270-6474/16/3612118-12$15.00/0.
Figures
References
-
- Batalha VL, Pego JM, Fontinha BM, Costenla AR, Valadas JS, Baqi Y, Radjainia H, Müller CE, Sebastião AM, Lopes LV. Adenosine A(2A) receptor blockade reverts hippocampal stress-induced deficits and restores corticosterone circadian oscillation. Mol Psychiatry. 2013;18:320–331. doi: 10.1038/mp.2012.8. - DOI - PubMed
-
- Bjursell MK, Blom HJ, Cayuela JA, Engvall ML, Lesko N, Balasubramaniam S, Brandberg G, Halldin M, Falkenberg M, Jakobs C, Smith D, Struys E, von Döbeln U, Gustafsson CM, Lundeberg J, Wedell A. Adenosine kinase deficiency disrupts the methionine cycle and causes hypermethioninemia, encephalopathy, and abnormal liver function. Am J Hum Genet. 2011;89:507–515. doi: 10.1016/j.ajhg.2011.09.004. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous