

Cure for thalassemia major - from allogeneic hematopoietic stem cell transplantation to gene therapy
- PMID: 27909215
- PMCID: PMC5286930
- DOI: 10.3324/haematol.2015.141200
Cure for thalassemia major - from allogeneic hematopoietic stem cell transplantation to gene therapy
Abstract
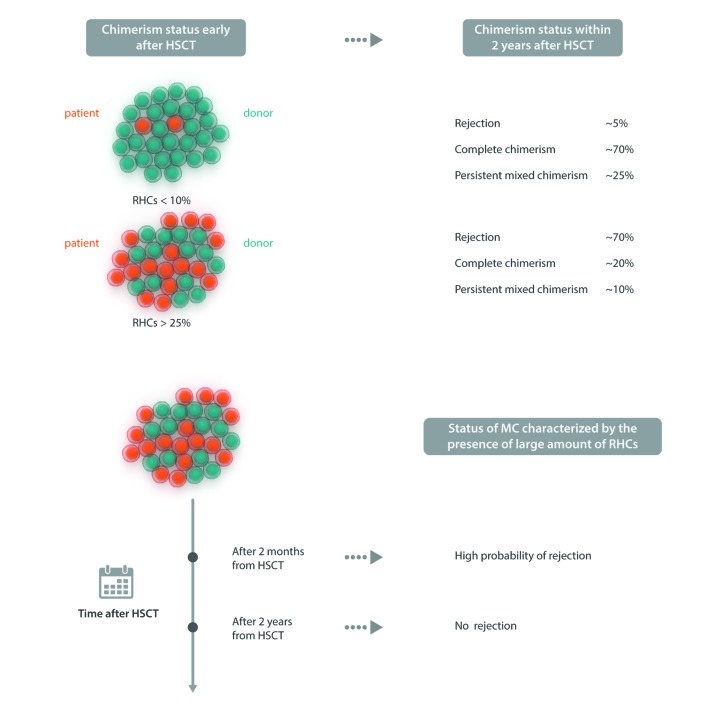
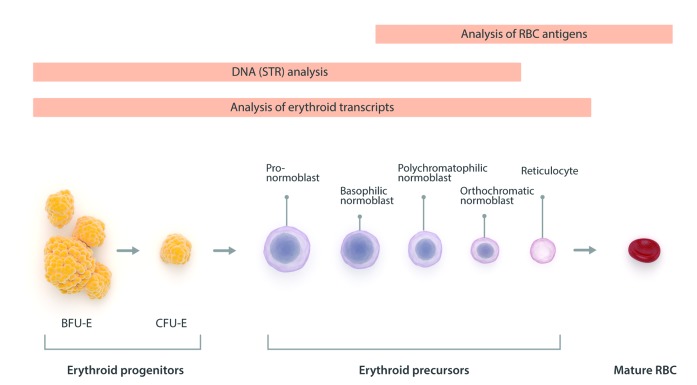
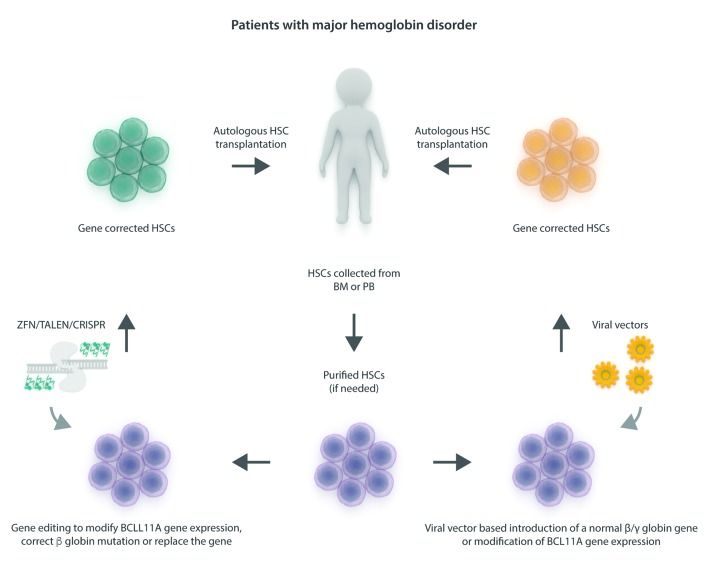
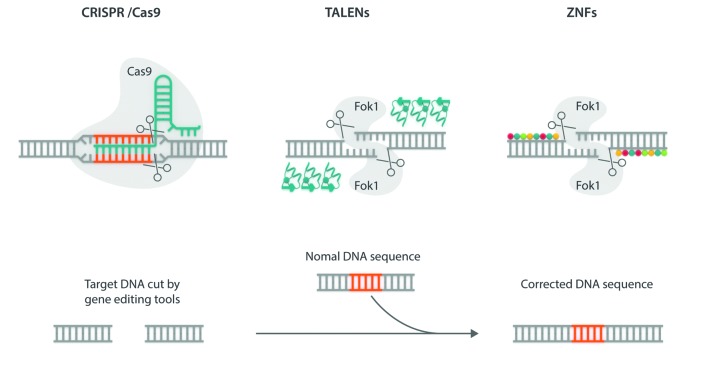
Allogeneic hematopoietic stem cell transplantation has been well established for several decades as gene replacement therapy for patients with thalassemia major, and now offers very high rates of cure for patients who have access to this therapy. Outcomes have improved tremendously over the last decade, even in high-risk patients. The limited data available suggests that the long-term outcome is also excellent, with a >90% survival rate, but for the best results, hematopoietic stem cell transplantation should be offered early, before any end organ damage occurs. However, access to this therapy is limited in more than half the patients by the lack of suitable donors. Inadequate hematopoietic stem cell transplantation services and the high cost of therapy are other reasons for this limited access, particularly in those parts of the world which have a high prevalence of this condition. As a result, fewer than 10% of eligible patients are actually able to avail of this therapy. Other options for curative therapies are therefore needed. Recently, gene correction of autologous hematopoietic stem cells has been successfully established using lentiviral vectors, and several clinical trials have been initiated. A gene editing approach to correct the β-globin mutation or disrupt the BCL11A gene to increase fetal hemoglobin production has also been reported, and is expected to be introduced in clinical trials soon. Curative possibilities for the major hemoglobin disorders are expanding. Providing access to these therapies around the world will remain a challenge.
Copyright© Ferrata Storti Foundation.
Figures
Similar articles
-
Gene Therapy and Genome Editing.Hematol Oncol Clin North Am. 2018 Apr;32(2):329-342. doi: 10.1016/j.hoc.2017.11.007. Epub 2018 Jan 9. Hematol Oncol Clin North Am. 2018. PMID: 29458735 Review.
-
A New Era for Hemoglobinopathies: More Than One Curative Option.Curr Gene Ther. 2017;17(5):364-378. doi: 10.2174/1566523218666180119123655. Curr Gene Ther. 2017. PMID: 29357790 Review.
-
Hematopoietic Stem Cell Gene-Addition/Editing Therapy in Sickle Cell Disease.Cells. 2022 Jun 4;11(11):1843. doi: 10.3390/cells11111843. Cells. 2022. PMID: 35681538 Free PMC article. Review.
-
Hurdles to the Adoption of Gene Therapy as a Curative Option for Transfusion-Dependent Thalassemia.Stem Cells Transl Med. 2022 Apr 29;11(4):407-414. doi: 10.1093/stcltm/szac007. Stem Cells Transl Med. 2022. PMID: 35267028 Free PMC article.
-
Hemoglobin gene therapy for β-thalassemia.Hematol Oncol Clin North Am. 2010 Dec;24(6):1187-201. doi: 10.1016/j.hoc.2010.08.002. Hematol Oncol Clin North Am. 2010. PMID: 21075288 Review.
Cited by
-
Screening of Some Indicators for Alpha-Thalassemia in Fujian Province of Southern China.Int J Gen Med. 2021 Oct 28;14:7329-7335. doi: 10.2147/IJGM.S338419. eCollection 2021. Int J Gen Med. 2021. PMID: 34737627 Free PMC article.
-
Is Old (Fludrabine/Busulfan/Cyclophosphamide/rAntiThymocyteGlobulin) Conditioning Still Gold for Allogeneic Transplants in Transfusion Dependent Beta-Thalassemia of All Risk Categories in 21st Century?Indian J Hematol Blood Transfus. 2023 Apr 3;39(4):1-9. doi: 10.1007/s12288-023-01646-1. Online ahead of print. Indian J Hematol Blood Transfus. 2023. PMID: 37362406 Free PMC article.
-
CRISPR-based therapeutic genome editing for inherited blood disorders.Nat Rev Drug Discov. 2025 Jul 14. doi: 10.1038/s41573-025-01236-y. Online ahead of print. Nat Rev Drug Discov. 2025. PMID: 40659814 Review.
-
Novel genetic therapeutic approaches for modulating the severity of β-thalassemia (Review).Biomed Rep. 2020 Nov;13(5):48. doi: 10.3892/br.2020.1355. Epub 2020 Sep 2. Biomed Rep. 2020. PMID: 32953110 Free PMC article. Review.
-
Busulfan-fludarabine- or treosulfan-fludarabine-based myeloablative conditioning for children with thalassemia major.Ann Hematol. 2022 Mar;101(3):655-665. doi: 10.1007/s00277-021-04732-4. Epub 2022 Jan 9. Ann Hematol. 2022. PMID: 34999929
References
-
- Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassaemia. Lancet. 2012;379(9813): 373–383. - PubMed
-
- Giardina PJ. THALASSEMIA SYNDROMES. In: Hoffman R, ed. Hematology: Basic Principles and Practice. 5th Edition ed. Philadelphia, PA: Elsevier, 2008:535–563.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
