

Deconstructing the Bat Skin Microbiome: Influences of the Host and the Environment
- PMID: 27909426
- PMCID: PMC5112243
- DOI: 10.3389/fmicb.2016.01753
Deconstructing the Bat Skin Microbiome: Influences of the Host and the Environment
Abstract
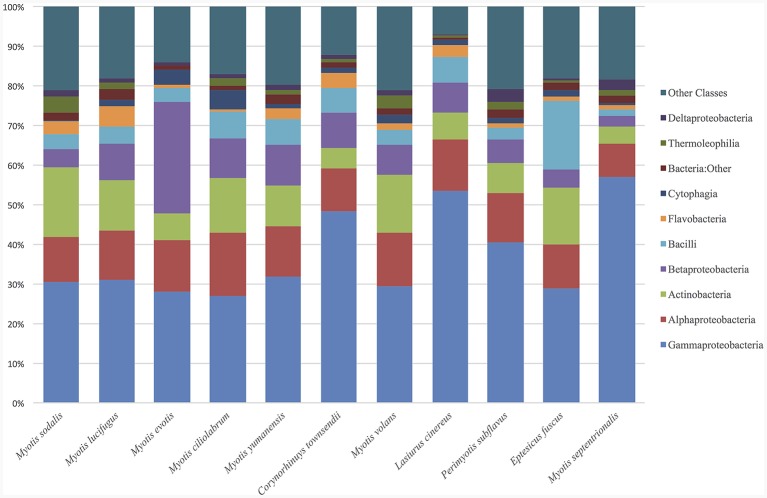
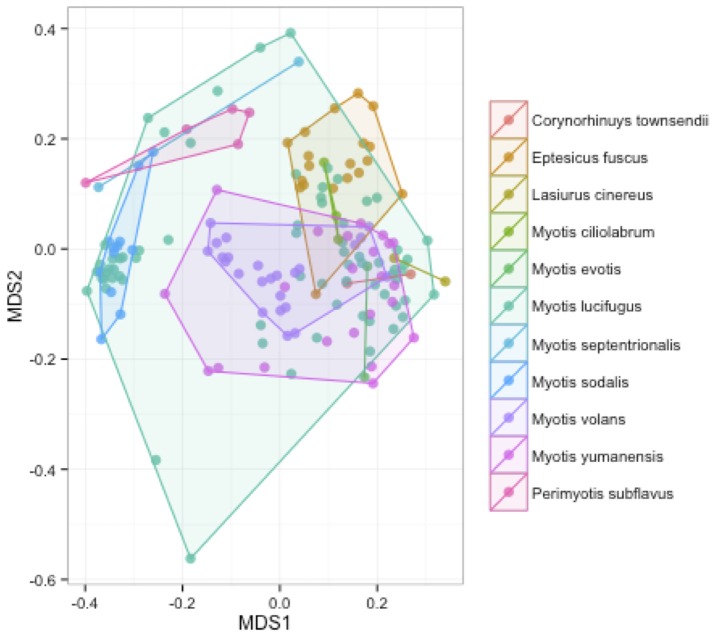
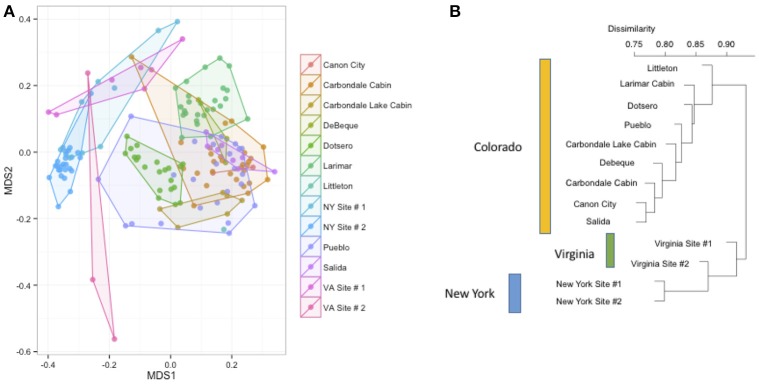
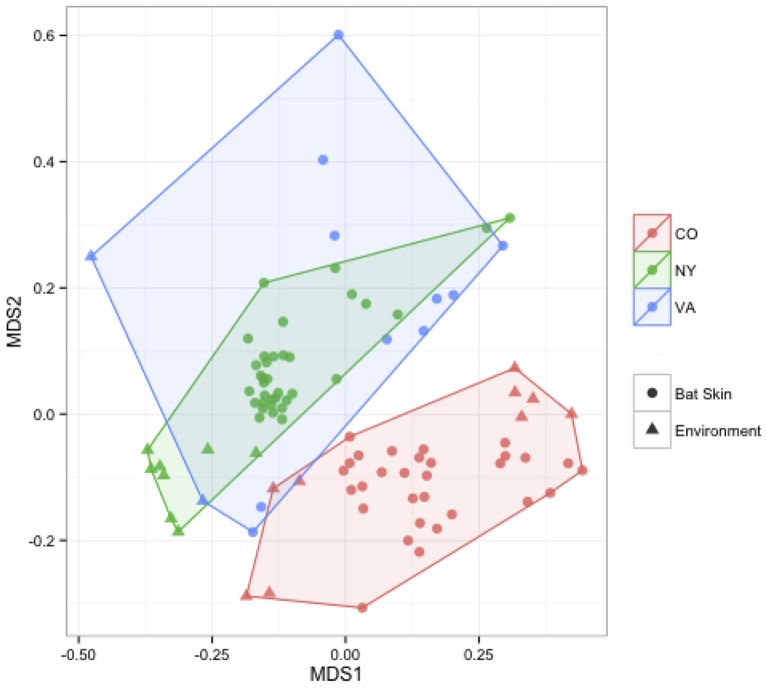
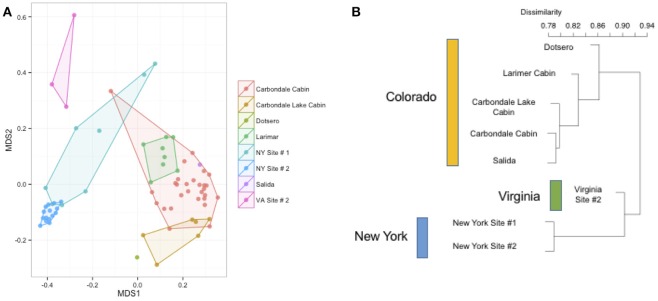
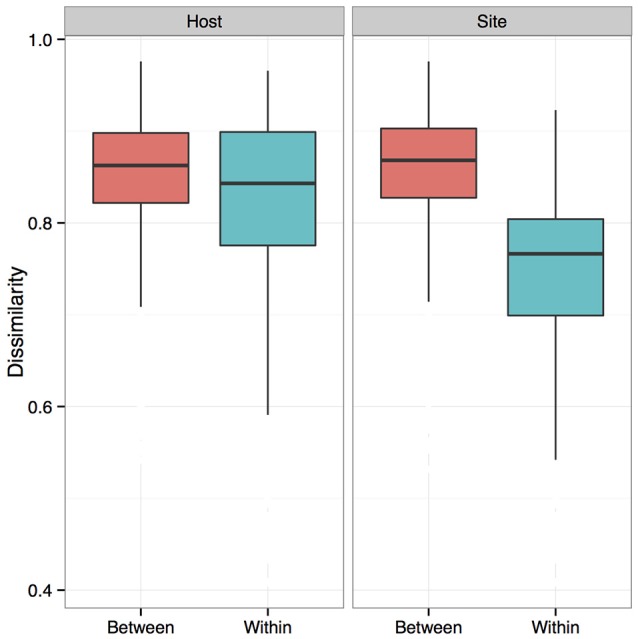
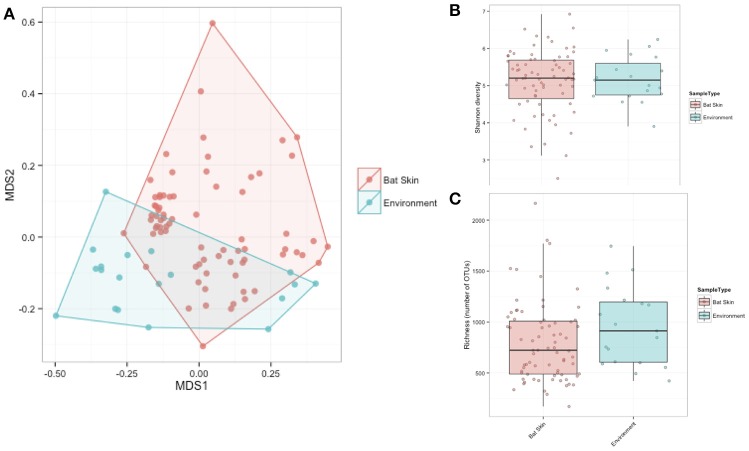
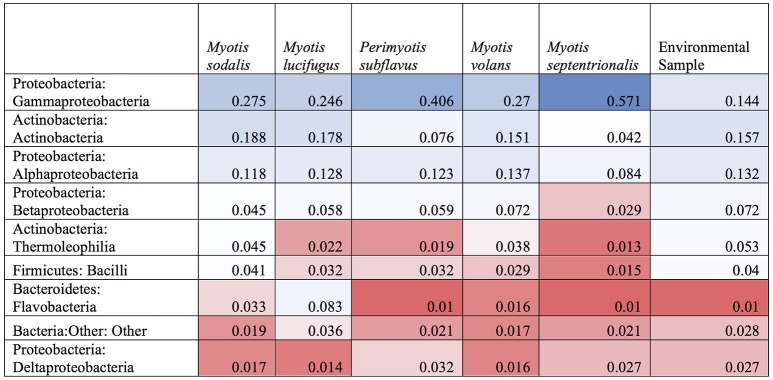
Bats are geographically widespread and play an important role in many ecosystems, but relatively little is known about the ecology of their associated microbial communities and the role microbial taxa play in bat health, development, and evolution. Moreover, few vertebrate animal skin microbiomes have been comprehensively assessed, and thus characterizing the bat skin microbiome will yield valuable insight into the variability of vertebrate skin microbiomes as a whole. The recent emergence of the skin fungal disease white-nose syndrome highlights the potentially important role bat skin microbial communities could play in bat health. Understanding the determinant of bat skin microbial communities could provide insight into important factors allowing individuals to persist with disease. We collected skin swabs from a total of 11 bat species from the eastern United States (n = 45) and Colorado (n = 119), as well as environmental samples (n = 38) from a subset of sites, and used 16S rRNA marker gene sequencing to observe bacterial communities. In addition, we conducted a literature survey to compare the skin microbiome across vertebrate groups, including the bats presented in this study. Host species, region, and site were all significant predictors of the variability across bat skin bacterial communities. Many bacterial taxa were found both on bats and in the environment. However, some bacterial taxa had consistently greater relative abundances on bat skin relative to their environments. Bats shared many of their abundant taxa with other vertebrates, but also hosted unique bacterial lineages such as the class Thermoleophilia (Actinobacteria). A strong effect of site on the bat skin microbiome indicates that the environment very strongly influences what bacteria are present on bat skin. Bat skin microbiomes are largely composed of site-specific microbiota, but there do appear to be important host-specific taxa. How this translates to differences in host-microbial interactions and bat health remains an important knowledge gap, but this work suggests that habitat variability is very important. We identify some bacterial groups that are more consistent on bats despite site differences, and these may be important ones to study in terms of their function as potential core microbiome members.
Keywords: 16S rRNA; bat ecology; host-associated bacteria; microbial ecology; microbiome; molecular ecology; white-nose syndrome.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous