

Beyond ferryl-mediated hydroxylation: 40 years of the rebound mechanism and C-H activation
- PMID: 27909920
- PMCID: PMC5350257
- DOI: 10.1007/s00775-016-1414-3
Beyond ferryl-mediated hydroxylation: 40 years of the rebound mechanism and C-H activation
Abstract
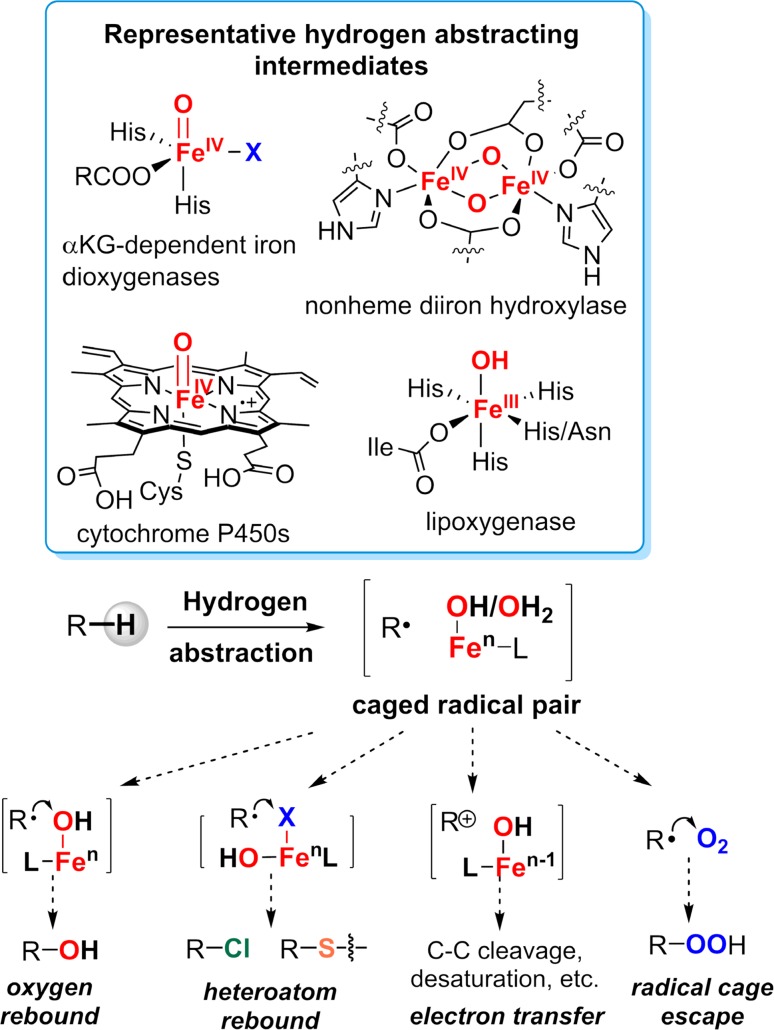
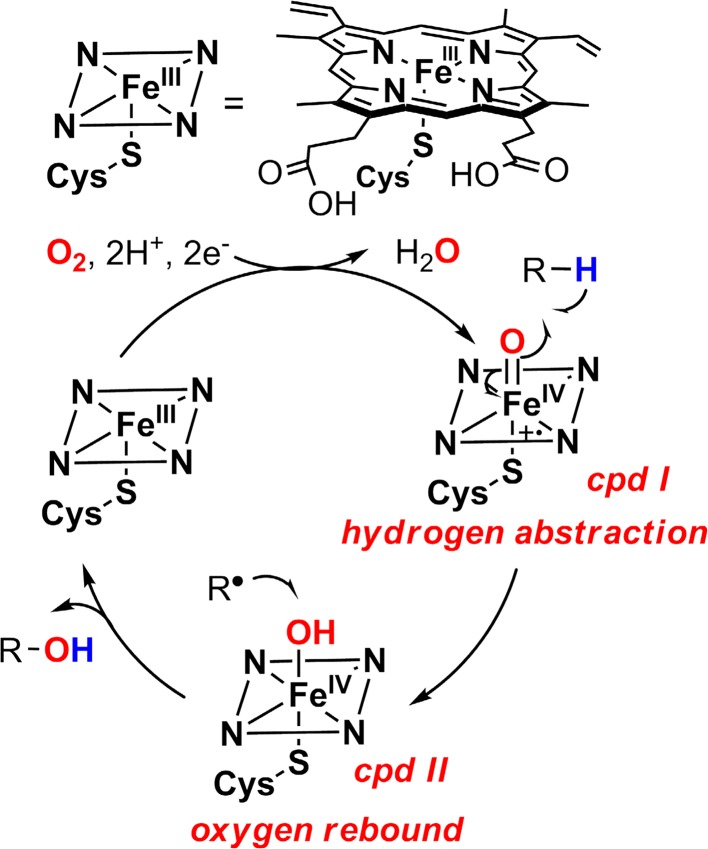
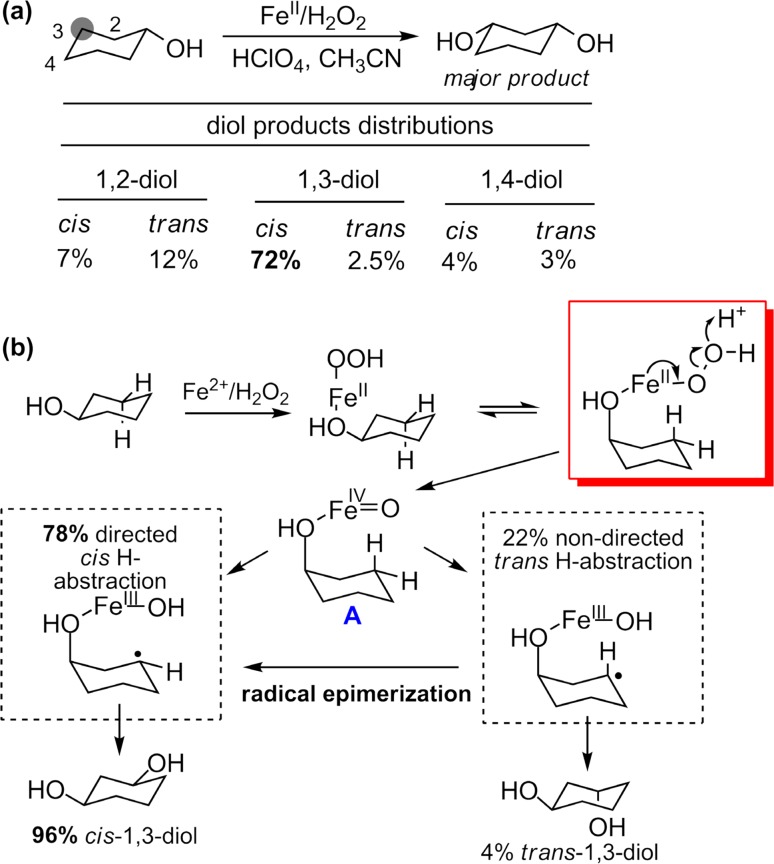
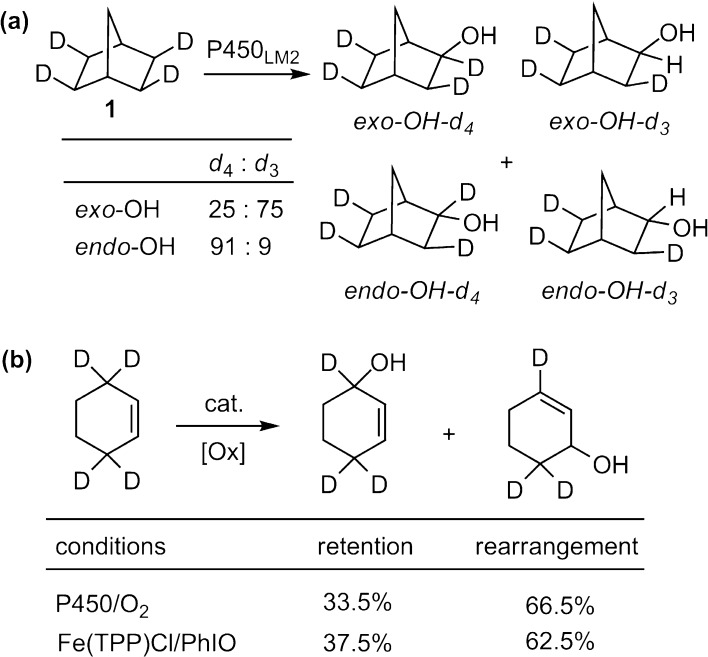
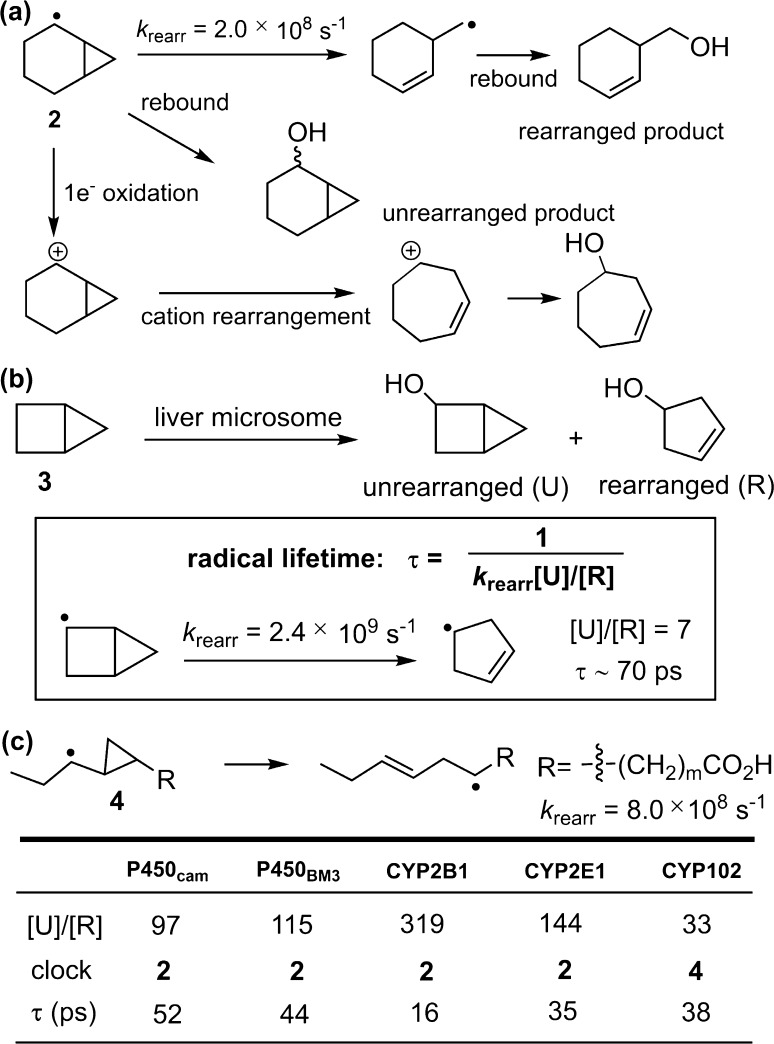
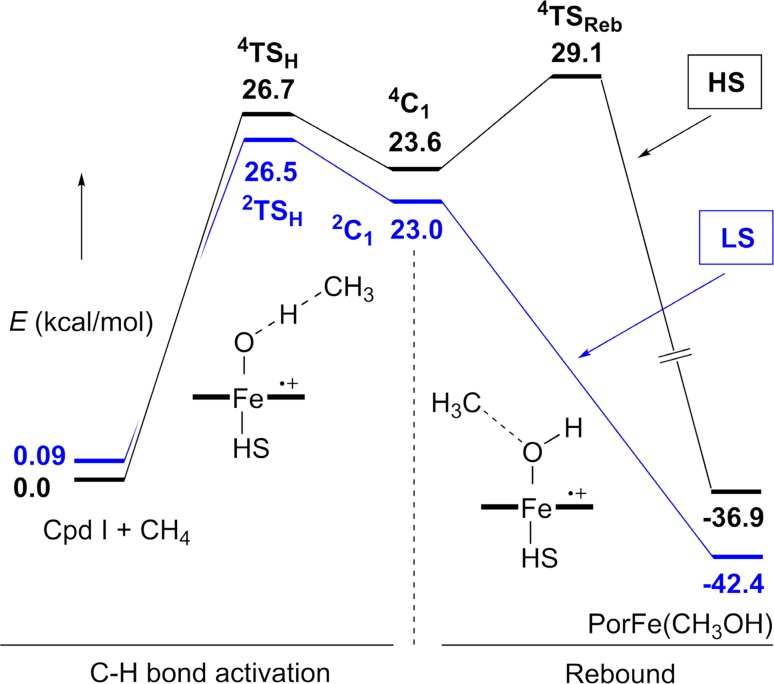
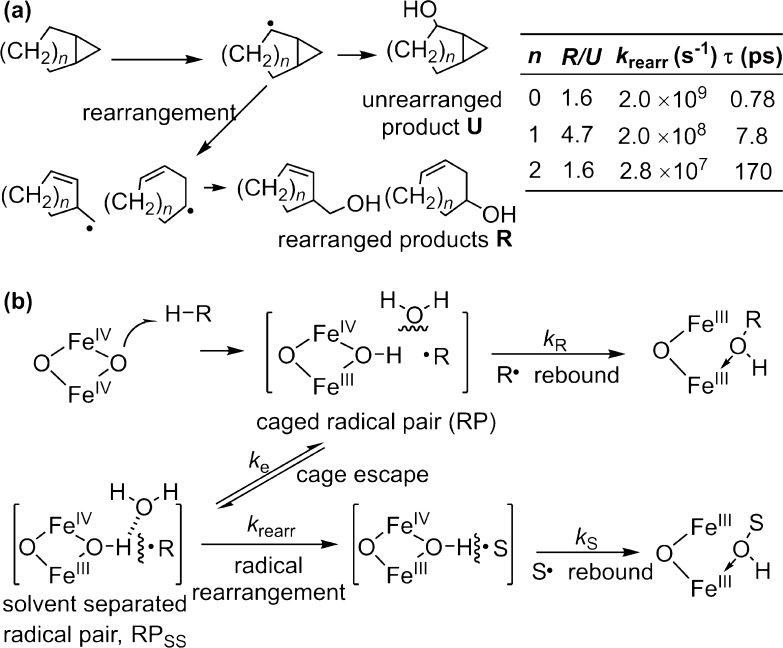
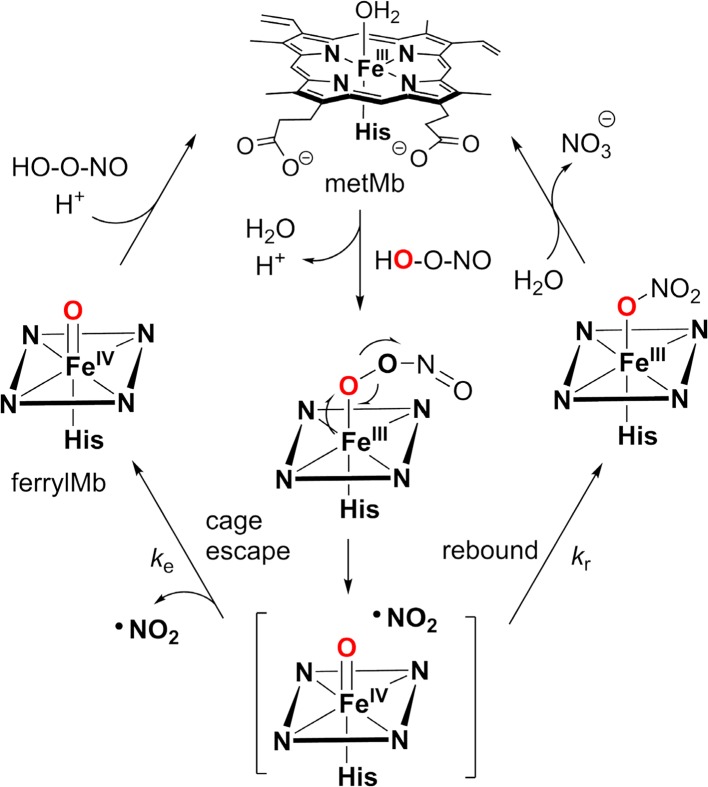
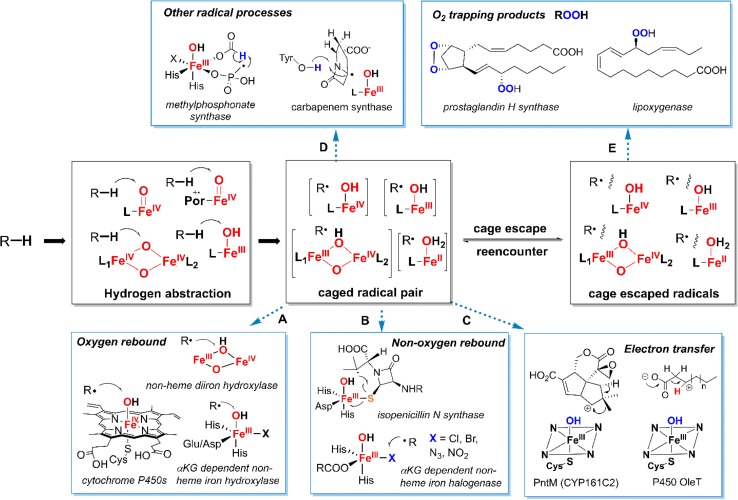
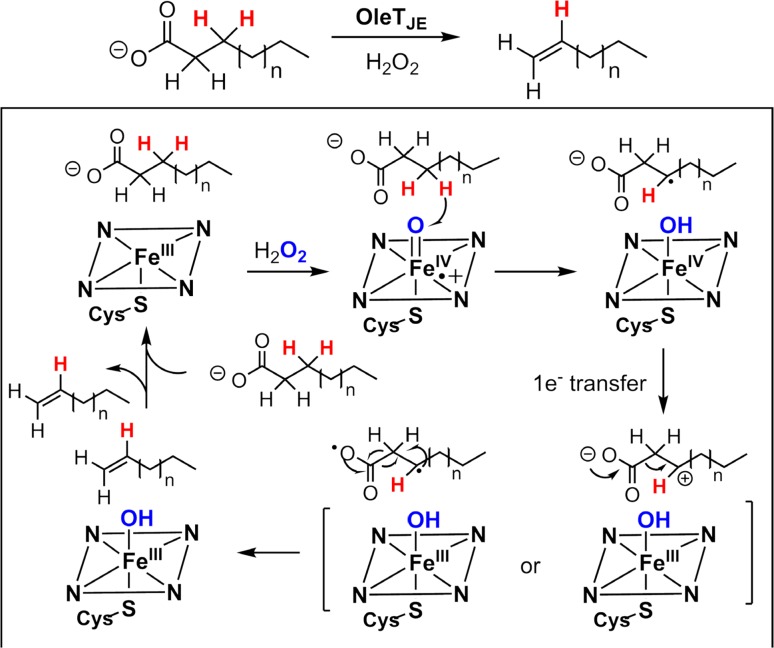
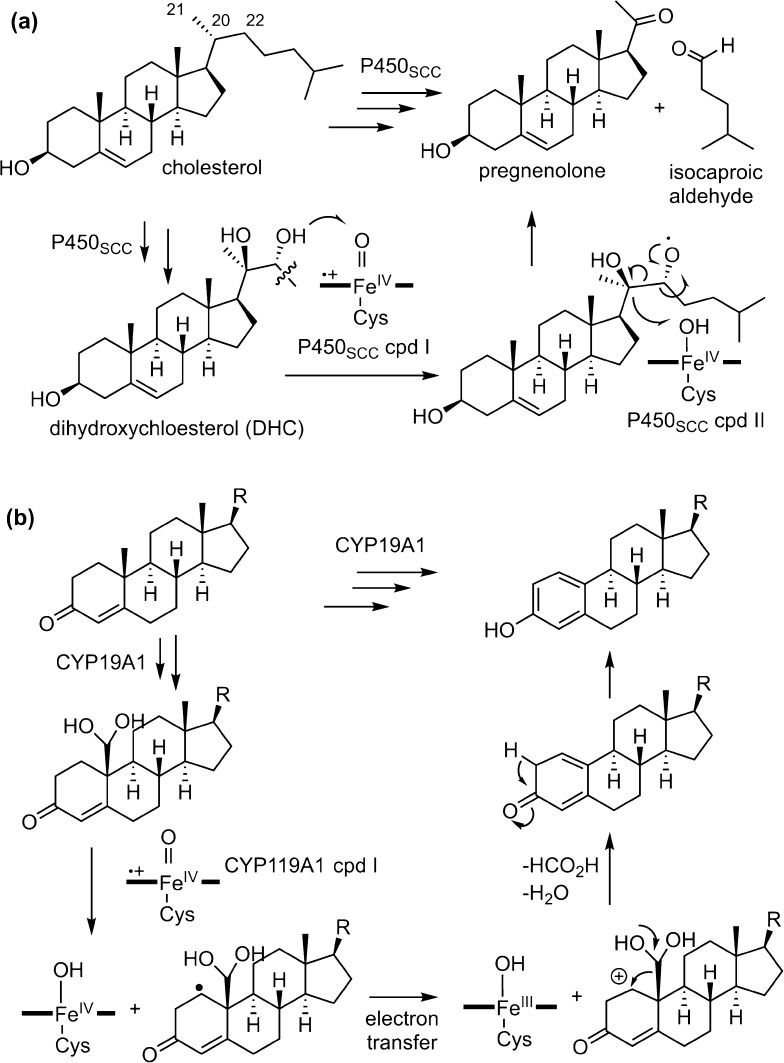
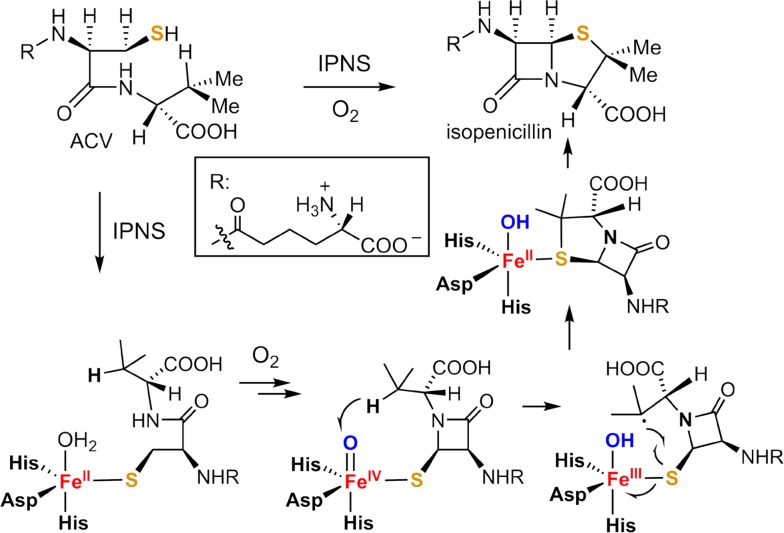
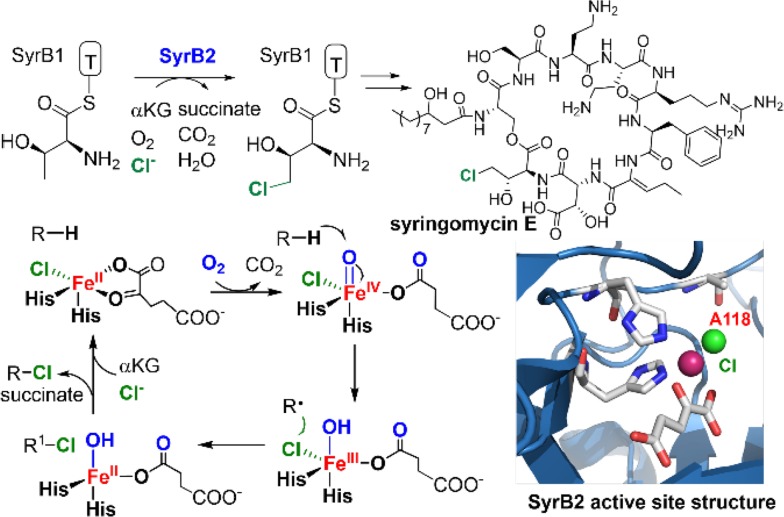
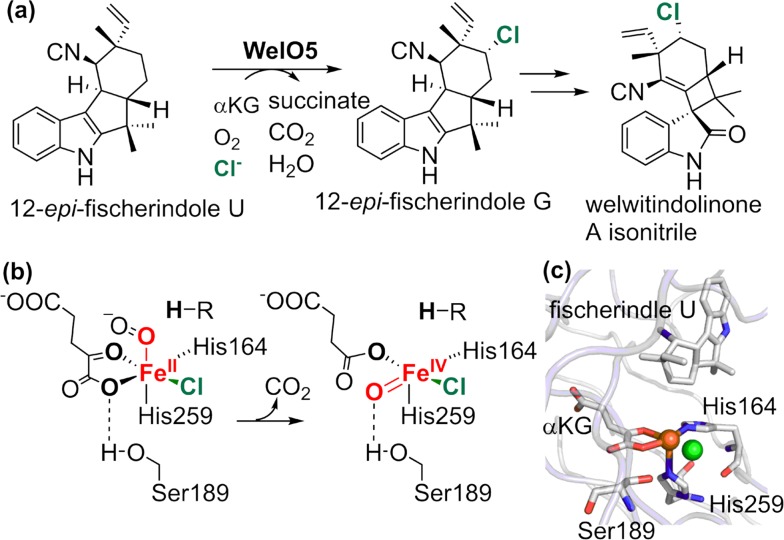
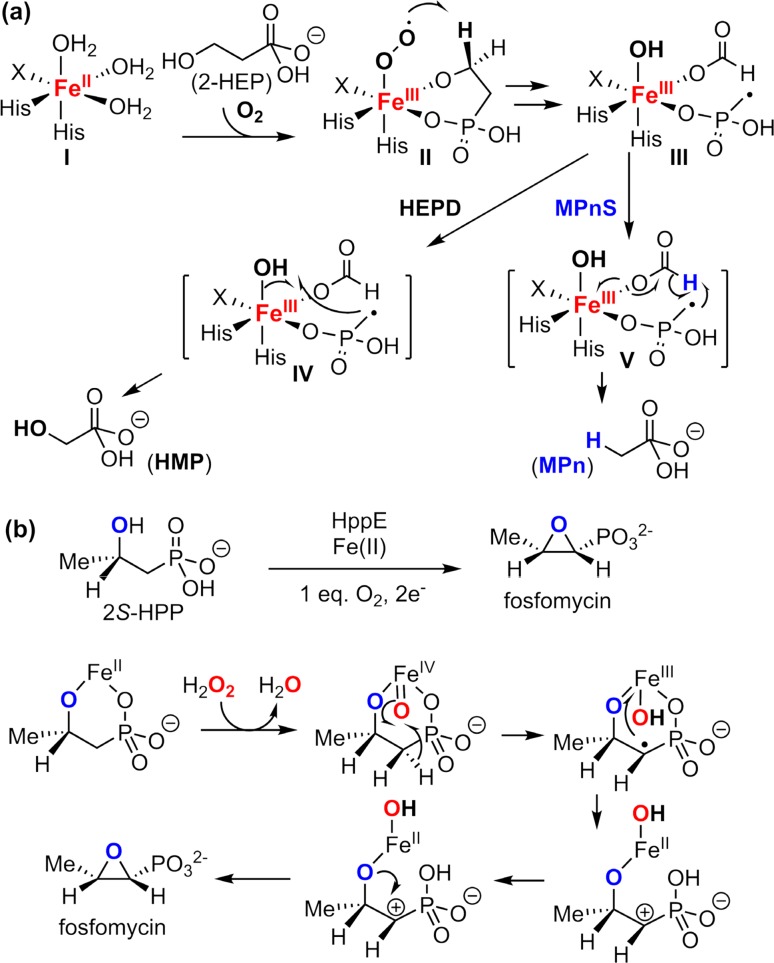
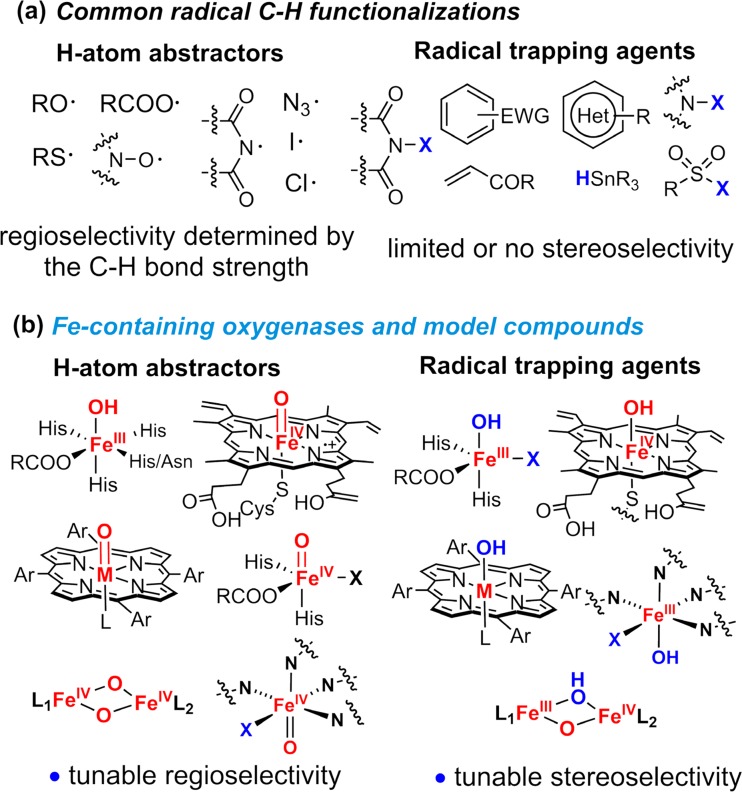
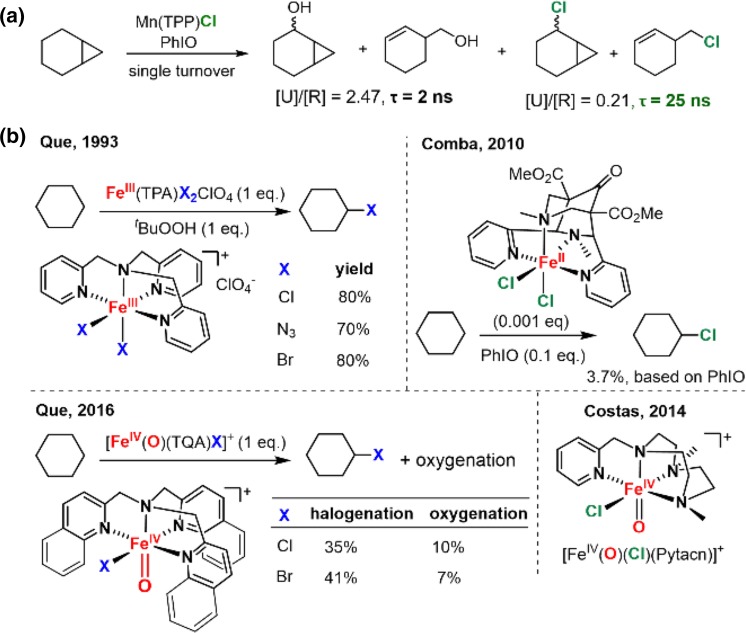
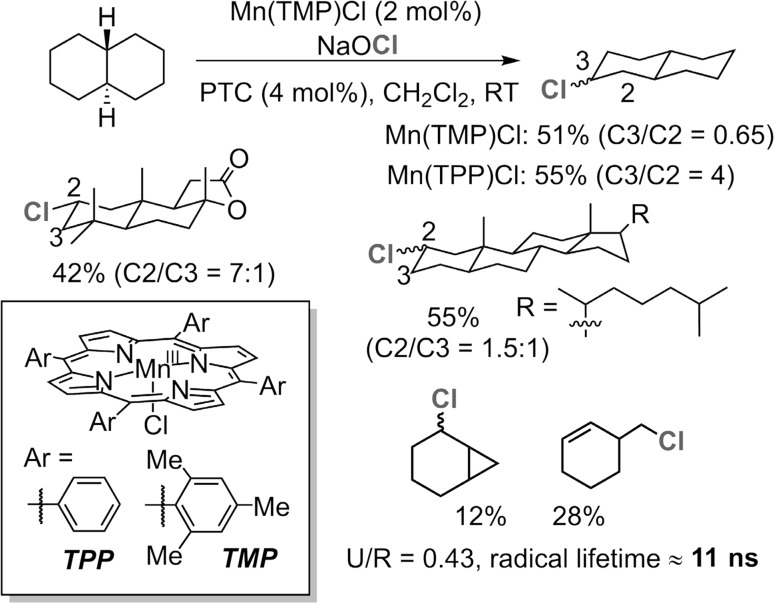
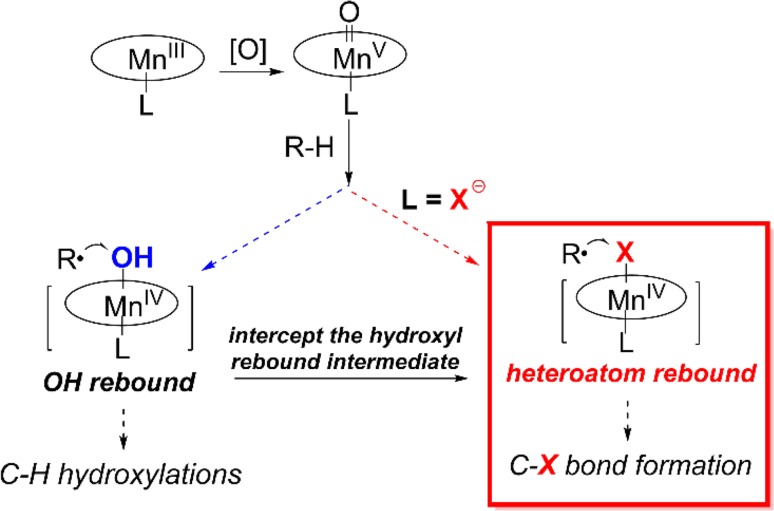
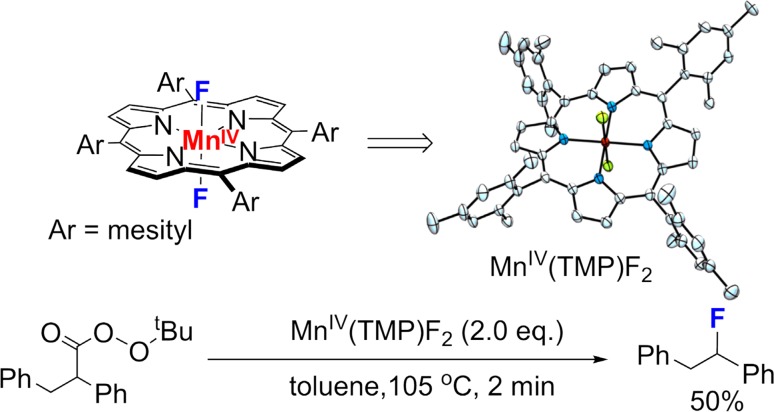
Since our initial report in 1976, the oxygen rebound mechanism has become the consensus mechanistic feature for an expanding variety of enzymatic C-H functionalization reactions and small molecule biomimetic catalysts. For both the biotransformations and models, an initial hydrogen atom abstraction from the substrate (R-H) by high-valent iron-oxo species (Fen=O) generates a substrate radical and a reduced iron hydroxide, [Fen-1-OH ·R]. This caged radical pair then evolves on a complicated energy landscape through a number of reaction pathways, such as oxygen rebound to form R-OH, rebound to a non-oxygen atom affording R-X, electron transfer of the incipient radical to yield a carbocation, R+, desaturation to form olefins, and radical cage escape. These various flavors of the rebound process, often in competition with each other, give rise to the wide range of C-H functionalization reactions performed by iron-containing oxygenases. In this review, we first recount the history of radical rebound mechanisms, their general features, and key intermediates involved. We will discuss in detail the factors that affect the behavior of the initial caged radical pair and the lifetimes of the incipient substrate radicals. Several representative examples of enzymatic C-H transformations are selected to illustrate how the behaviors of the radical pair [Fen-1-OH ·R] determine the eventual reaction outcome. Finally, we discuss the powerful potential of "radical rebound" processes as a general paradigm for developing novel C-H functionalization reactions with synthetic, biomimetic catalysts. We envision that new chemistry will continue to arise by bridging enzymatic "radical rebound" with synthetic organic chemistry.
Keywords: C–H activation; Iron; Metal oxo; Oxygenase; Radical; Rebound.
Figures
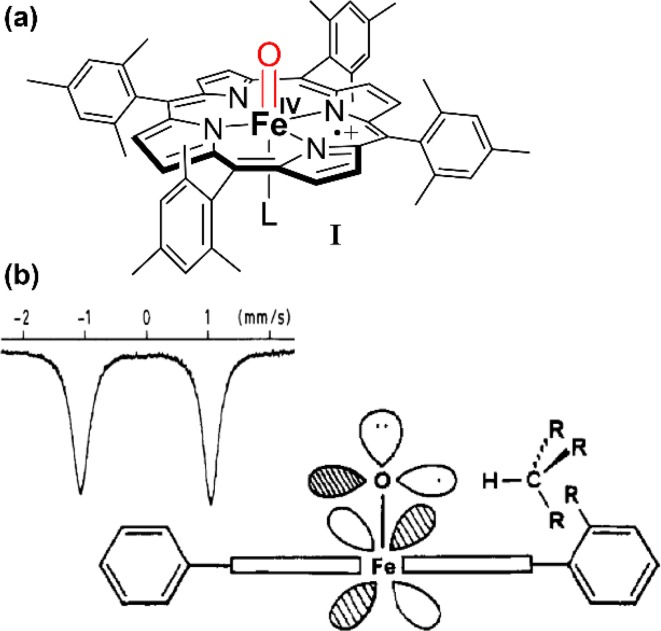
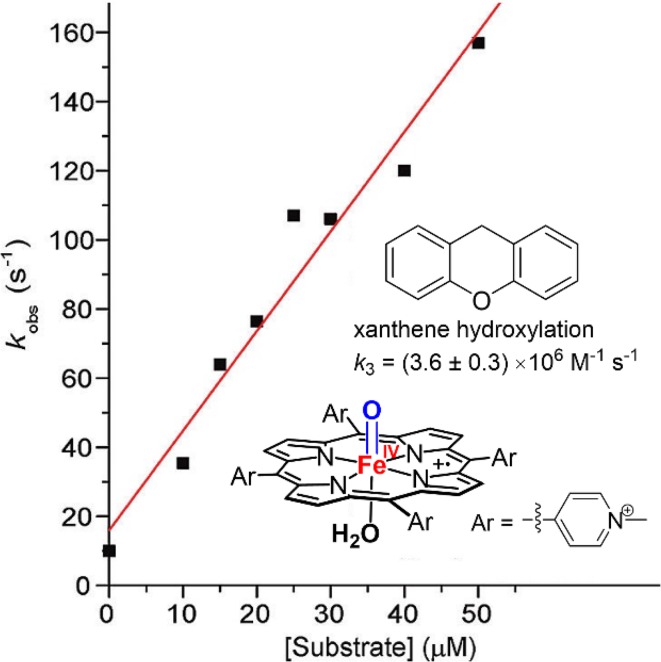
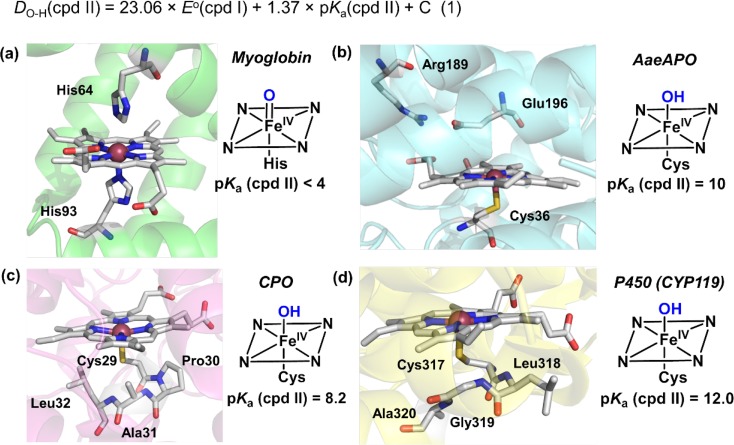
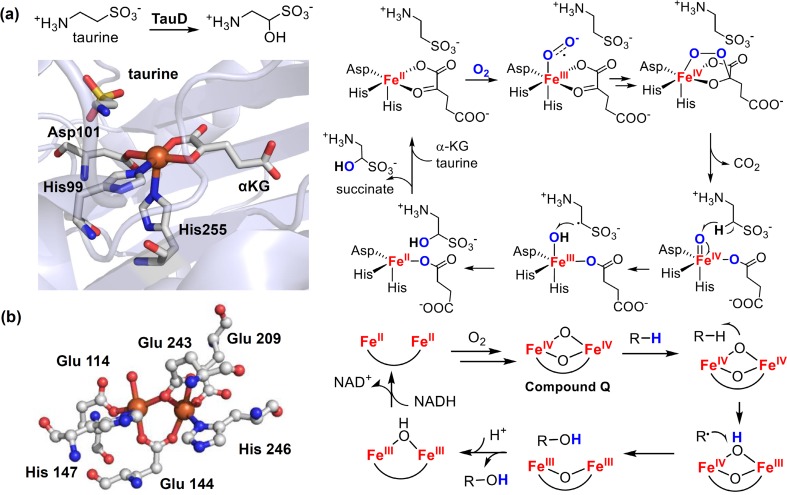

Similar articles
-
Evidence for an alternative to the oxygen rebound mechanism in C-H bond activation by non-heme Fe(IV)O complexes.J Am Chem Soc. 2012 Dec 19;134(50):20222-5. doi: 10.1021/ja308290r. Epub 2012 Dec 6. J Am Chem Soc. 2012. PMID: 23205855
-
The "somersault" mechanism for the p-450 hydroxylation of hydrocarbons. The intervention of transient inverted metastable hydroperoxides.J Am Chem Soc. 2006 Feb 8;128(5):1474-88. doi: 10.1021/ja052111+. J Am Chem Soc. 2006. PMID: 16448118
-
Is the ruthenium analogue of compound I of cytochrome p450 an efficient oxidant? A theoretical investigation of the methane hydroxylation reaction.J Am Chem Soc. 2003 Feb 26;125(8):2291-300. doi: 10.1021/ja0282487. J Am Chem Soc. 2003. PMID: 12590559
-
To rebound or dissociate? This is the mechanistic question in C-H hydroxylation by heme and nonheme metal-oxo complexes.Chem Soc Rev. 2016 Mar 7;45(5):1197-210. doi: 10.1039/c5cs00566c. Chem Soc Rev. 2016. PMID: 26690848 Review.
-
Models and mechanisms of O-O bond activation by cytochrome P450. A critical assessment of the potential role of multiple active intermediates in oxidative catalysis.Eur J Biochem. 2004 Nov;271(22):4335-60. doi: 10.1111/j.1432-1033.2004.04380.x. Eur J Biochem. 2004. PMID: 15560776 Review.
Cited by
-
Investigating the Active Oxidants Involved in Cytochrome P450 Catalyzed Sulfoxidation Reactions.Chemistry. 2022 Dec 27;28(72):e202202428. doi: 10.1002/chem.202202428. Epub 2022 Nov 9. Chemistry. 2022. PMID: 36169207 Free PMC article.
-
Oriented External Electric Fields Regurating the Reaction Mechanism of CH4 Oxidation Catalyzed by Fe(IV)-Oxo-Corrolazine: Insight from Density Functional Calculations.Front Chem. 2022 Jun 29;10:896944. doi: 10.3389/fchem.2022.896944. eCollection 2022. Front Chem. 2022. PMID: 35844657 Free PMC article.
-
Iron-Catalyzed Aerobic Carbonylation of Methane via Ligand-to-Metal Charge Transfer Excitation.J Am Chem Soc. 2025 Jan 15;147(2):1440-1447. doi: 10.1021/jacs.4c16449. Epub 2025 Jan 6. J Am Chem Soc. 2025. PMID: 39760382 Free PMC article.
-
Stereospecific Enzymatic Conversion of Boronic Acids to Amines.J Am Chem Soc. 2024 Jul 17;146(28):19160-19167. doi: 10.1021/jacs.4c04190. Epub 2024 Jul 3. J Am Chem Soc. 2024. PMID: 38958264 Free PMC article.
-
Hydrogen Atom Abstraction by High-Valent Fe(OH) versus Mn(OH) Porphyrinoid Complexes: Mechanistic Insights from Experimental and Computational Studies.Inorg Chem. 2019 Dec 16;58(24):16761-16770. doi: 10.1021/acs.inorgchem.9b02923. Epub 2019 Dec 5. Inorg Chem. 2019. PMID: 31804814 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
