

A comprehensive map of molecular drug targets
- PMID: 27910877
- PMCID: PMC6314433
- DOI: 10.1038/nrd.2016.230
A comprehensive map of molecular drug targets
Abstract
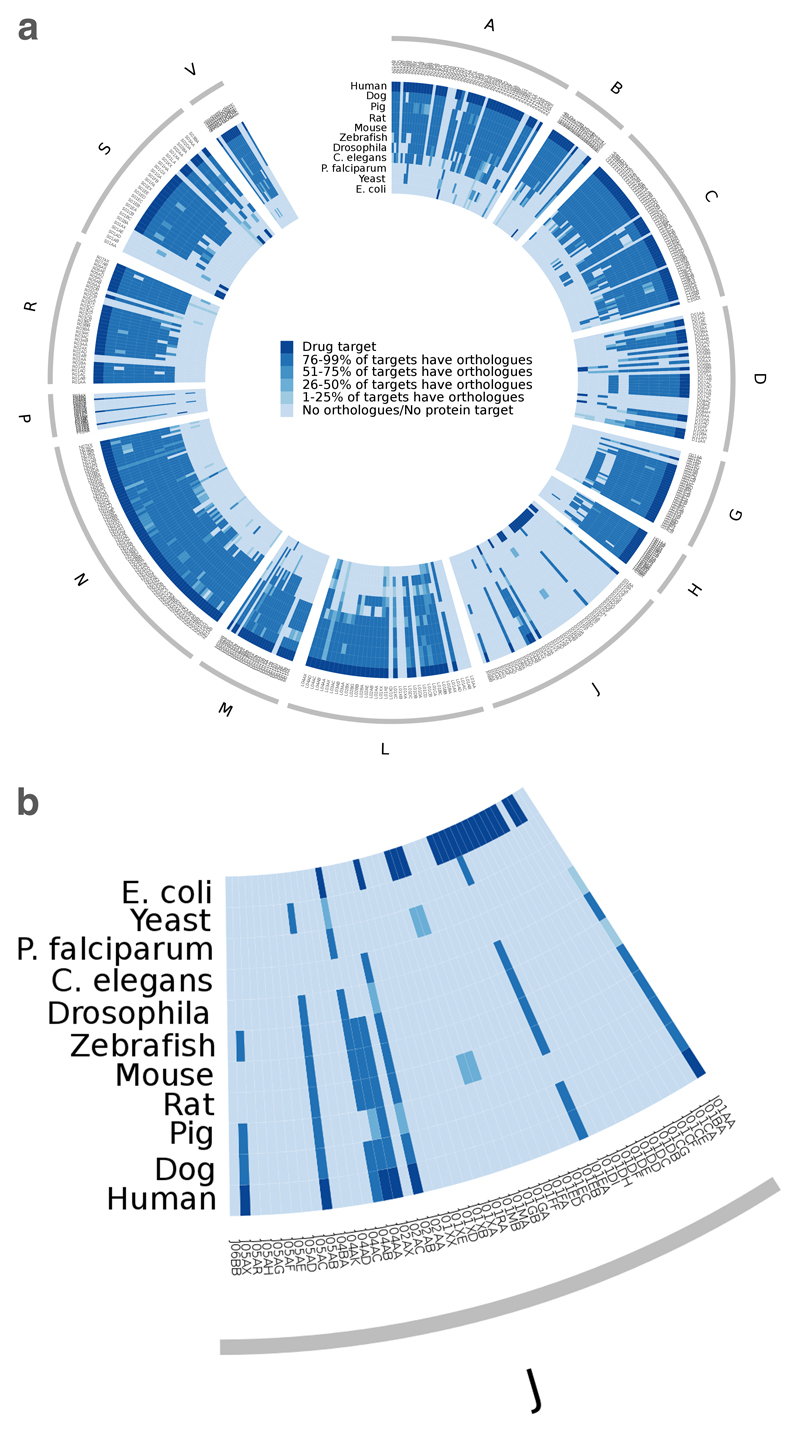
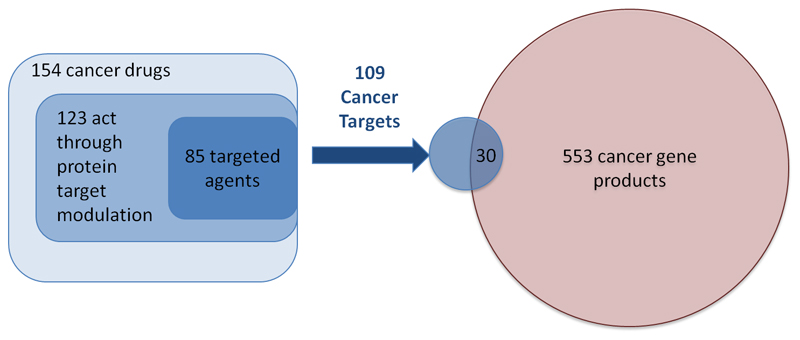
The success of mechanism-based drug discovery depends on the definition of the drug target. This definition becomes even more important as we try to link drug response to genetic variation, understand stratified clinical efficacy and safety, rationalize the differences between drugs in the same therapeutic class and predict drug utility in patient subgroups. However, drug targets are often poorly defined in the literature, both for launched drugs and for potential therapeutic agents in discovery and development. Here, we present an updated comprehensive map of molecular targets of approved drugs. We curate a total of 893 human and pathogen-derived biomolecules through which 1,578 US FDA-approved drugs act. These biomolecules include 667 human-genome-derived proteins targeted by drugs for human disease. Analysis of these drug targets indicates the continued dominance of privileged target families across disease areas, but also the growth of novel first-in-class mechanisms, particularly in oncology. We explore the relationships between bioactivity class and clinical success, as well as the presence of orthologues between human and animal models and between pathogen and human genomes. Through the collaboration of three independent teams, we highlight some of the ongoing challenges in accurately defining the targets of molecular therapeutics and present conventions for deconvoluting the complexities of molecular pharmacology and drug efficacy.
Conflict of interest statement
The authors declare no competing interests.
Figures
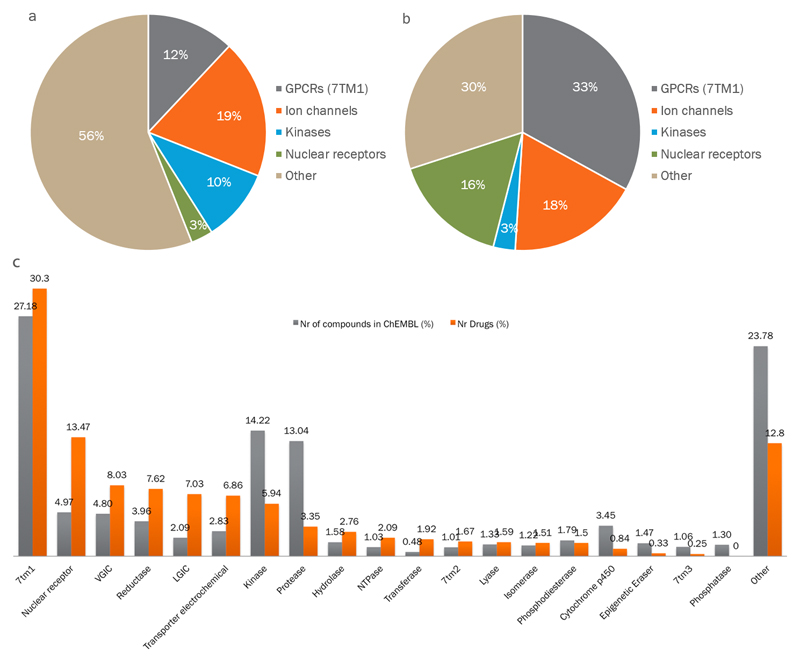
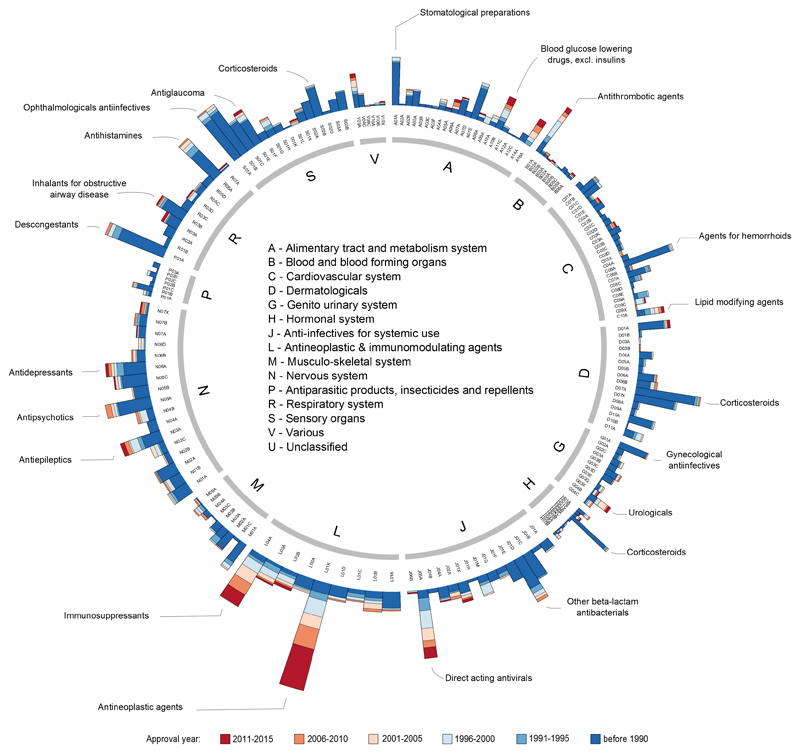
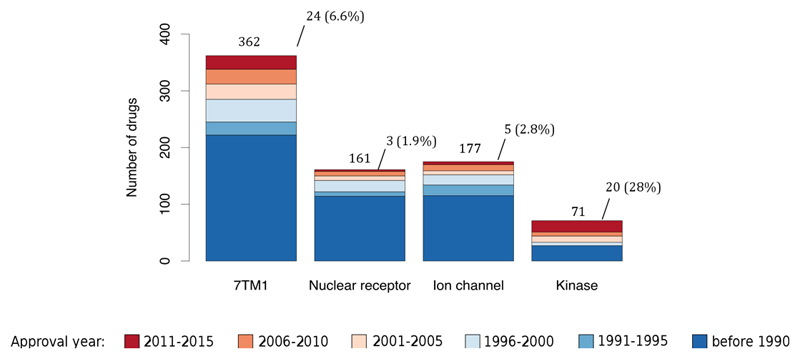
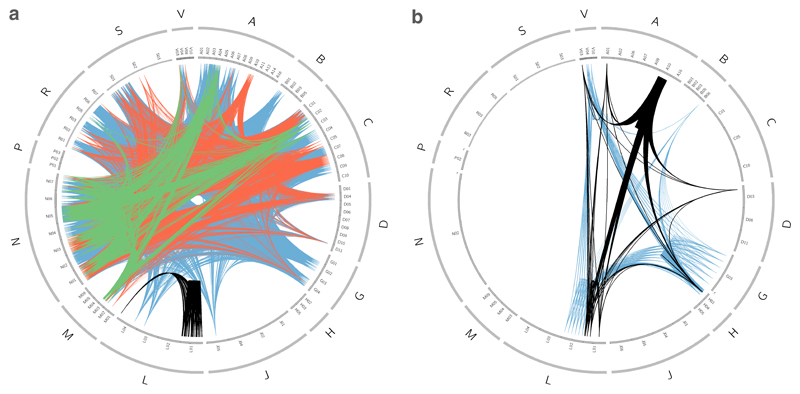
Comment in
-
Novel drug targets in 2023.Nat Rev Drug Discov. 2024 May;23(5):330. doi: 10.1038/d41573-024-00057-9. Nat Rev Drug Discov. 2024. PMID: 38565953 No abstract available.
References
-
- Raju TN. The Nobel chronicles. Lancet. 2000;355:1022. - PubMed
-
- Drews J. Genomic sciences and the medicine of tomorrow. Nat Biotechnol. 1996;14:1516–1518. [An early and influential review on the prospects for genomics and drug discovery.] - PubMed
-
- Drews J, Ryser S. The role of innovation in drug development. Nat Biotechnol. 1997;15:1318–1319. - PubMed
-
- Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–30. [First attempt to define the future drugabble genome on the basis of successful drug development programs.] - PubMed
-
- Golden JB. Prioritizing the human genome: knowledge management for drug discovery. Curr Opin Drug Discov Devel. 2003;6:310–316. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
