

Phosphatidylinositol 3-kinase inhibition restores Ca2+ release defects and prolongs survival in myotubularin-deficient mice
- PMID: 27911767
- PMCID: PMC5167204
- DOI: 10.1073/pnas.1604099113
Phosphatidylinositol 3-kinase inhibition restores Ca2+ release defects and prolongs survival in myotubularin-deficient mice
Abstract
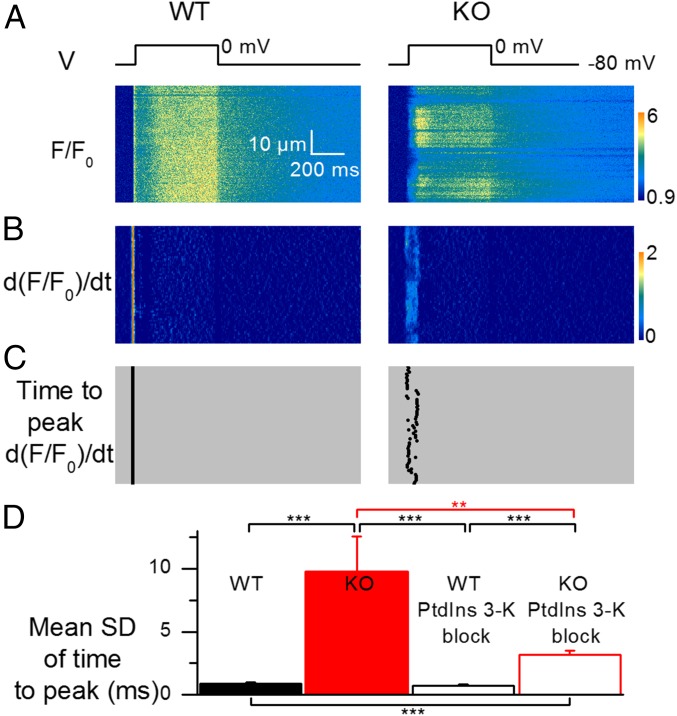
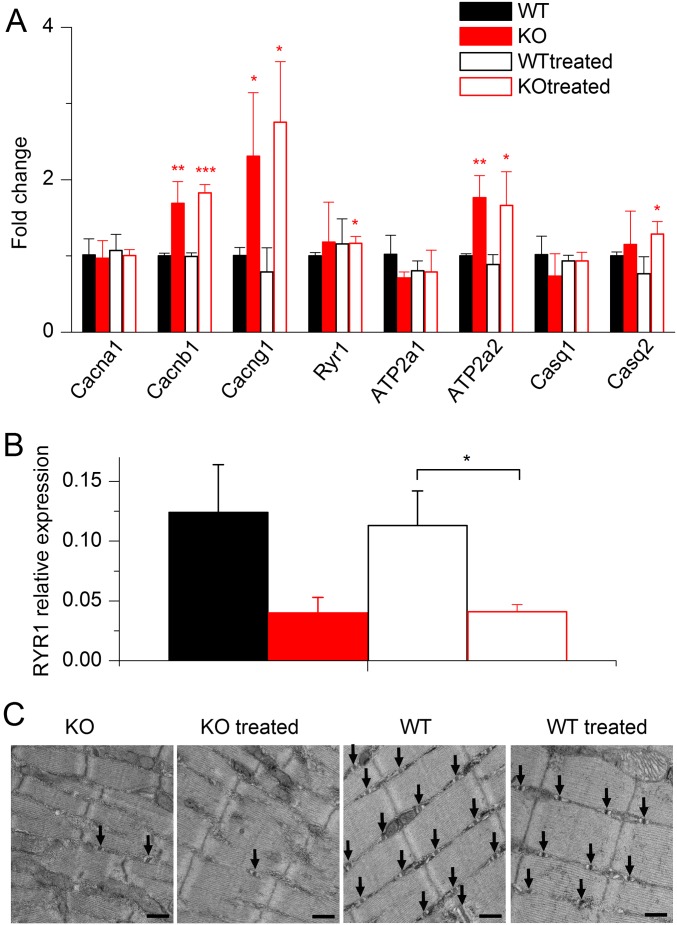
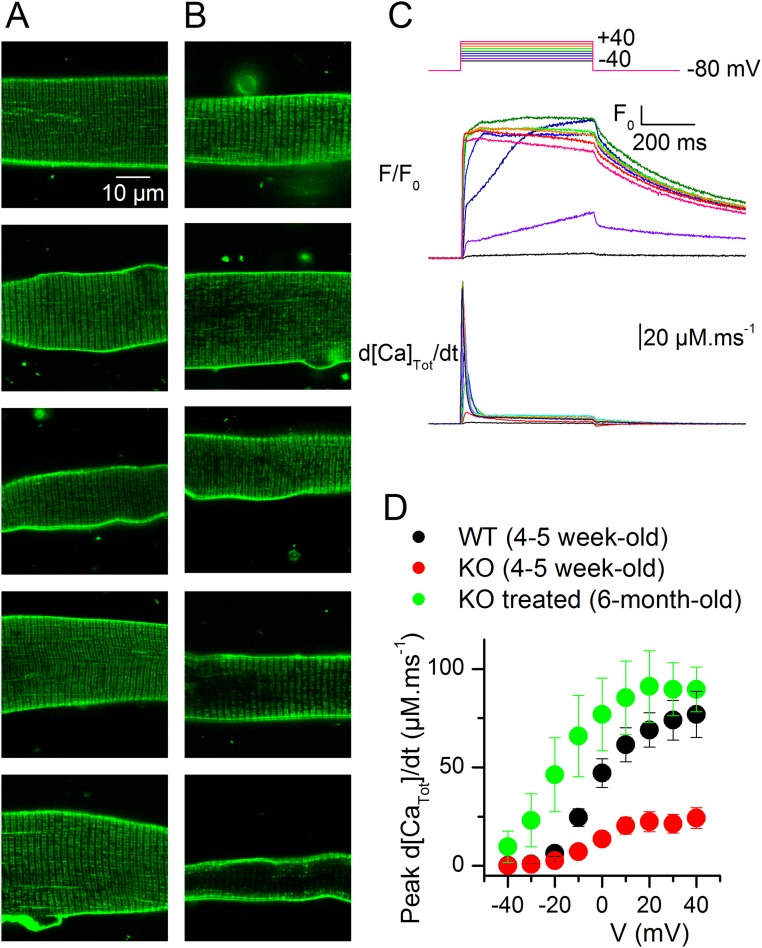
Mutations in the gene encoding the phosphoinositide 3-phosphatase myotubularin (MTM1) are responsible for a pediatric disease of skeletal muscle named myotubular myopathy (XLMTM). Muscle fibers from MTM1-deficient mice present defects in excitation-contraction (EC) coupling likely responsible for the disease-associated fatal muscle weakness. However, the mechanism leading to EC coupling failure remains unclear. During normal skeletal muscle EC coupling, transverse (t) tubule depolarization triggers sarcoplasmic reticulum (SR) Ca2+ release through ryanodine receptor channels gated by conformational coupling with the t-tubule voltage-sensing dihydropyridine receptors. We report that MTM1 deficiency is associated with a 60% depression of global SR Ca2+ release over the full range of voltage sensitivity of EC coupling. SR Ca2+ release in the diseased fibers is also slower than in normal fibers, or delayed following voltage activation, consistent with the contribution of Ca2+-gated ryanodine receptors to EC coupling. In addition, we found that SR Ca2+ release is spatially heterogeneous within myotubularin-deficient muscle fibers, with focally defective areas recapitulating the global alterations. Importantly, we found that pharmacological inhibition of phosphatidylinositol 3-kinase (PtdIns 3-kinase) activity rescues the Ca2+ release defects in isolated muscle fibers and increases the lifespan and mobility of XLMTM mice, providing proof of concept for the use of PtdIns 3-kinase inhibitors in myotubular myopathy and suggesting that unbalanced PtdIns 3-kinase activity plays a critical role in the pathological process.
Keywords: excitation–contraction coupling; myotubularin; ryanodine receptor; sarcoplasmic reticulum Ca2+ release; skeletal muscle.
Conflict of interest statement
M.W.L. receives research funding support from Audentes Therapeutics, Solid GT, and Demeter Therapeutics; is a member of the Scientific Advisory Board of Audentes Therapeutics; and was recently a consultant for Sarepta Therapeutics. A.B.-B. is a scientific advisor of Audentes Therapeutics.
Figures









References
-
- Ríos E, Pizarro G, Stefani E. Charge movement and the nature of signal transduction in skeletal muscle excitation-contraction coupling. Annu Rev Physiol. 1992;54:109–133. - PubMed
-
- Schneider M-F. Control of calcium release in functioning skeletal muscle fibers. Annu Rev Physiol. 1994;56:463–484. - PubMed
-
- Tronchère H, et al. Production of phosphatidylinositol 5-phosphate by the phosphoinositide 3-phosphatase myotubularin in mammalian cells. J Biol Chem. 2004;279(8):7304–7312. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
