

Selective sweep suggests transcriptional regulation may underlie Plasmodium vivax resilience to malaria control measures in Cambodia
- PMID: 27911780
- PMCID: PMC5167194
- DOI: 10.1073/pnas.1608828113
Selective sweep suggests transcriptional regulation may underlie Plasmodium vivax resilience to malaria control measures in Cambodia
Abstract
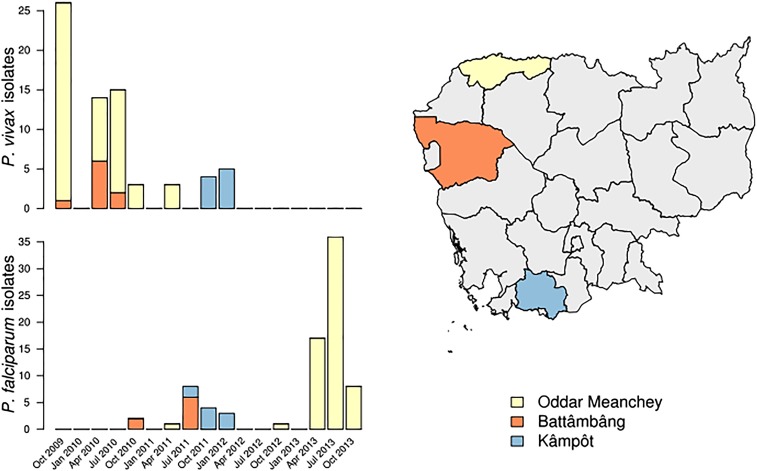
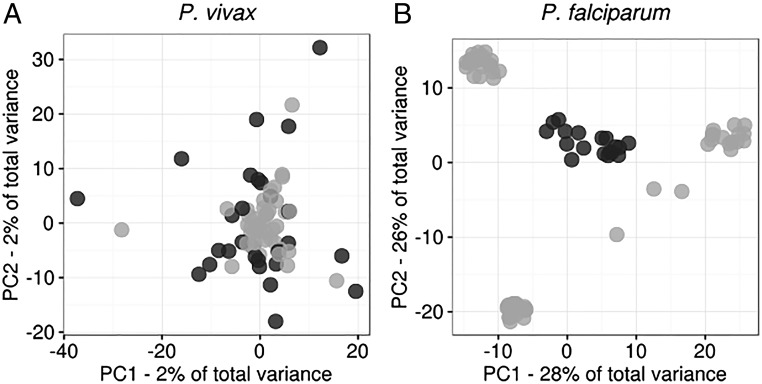
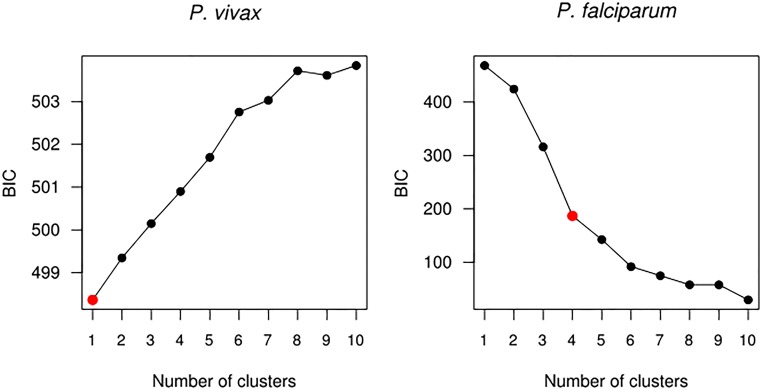
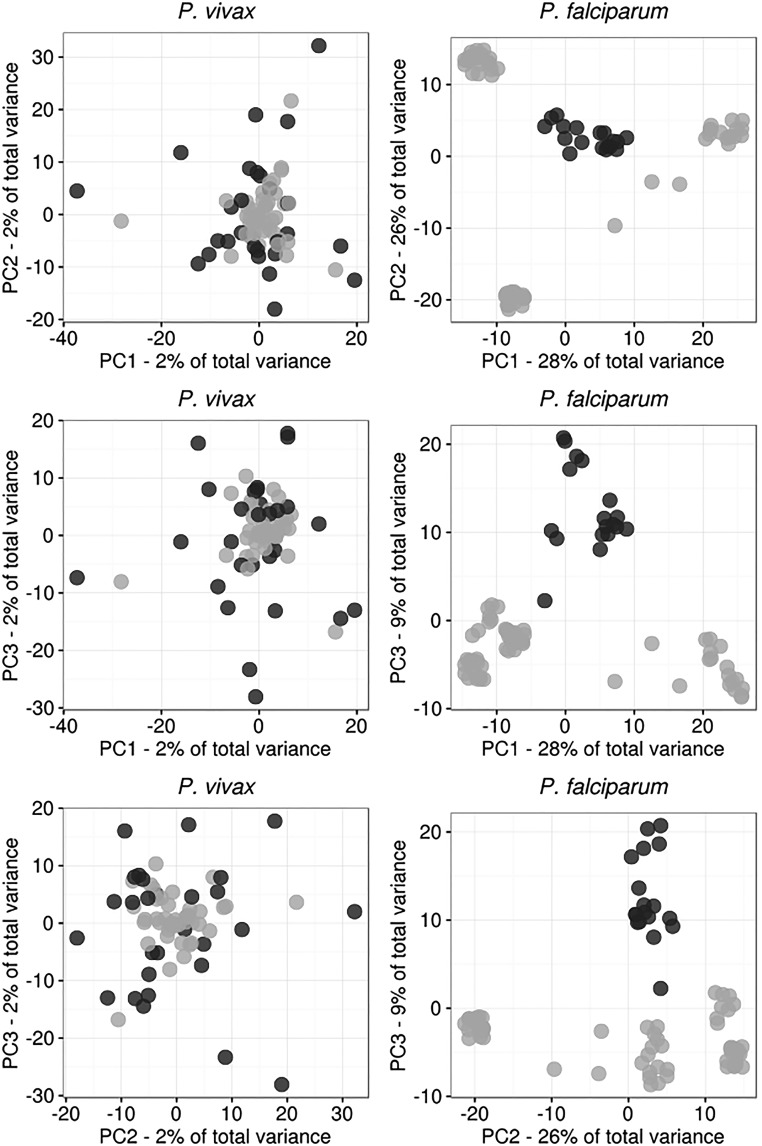
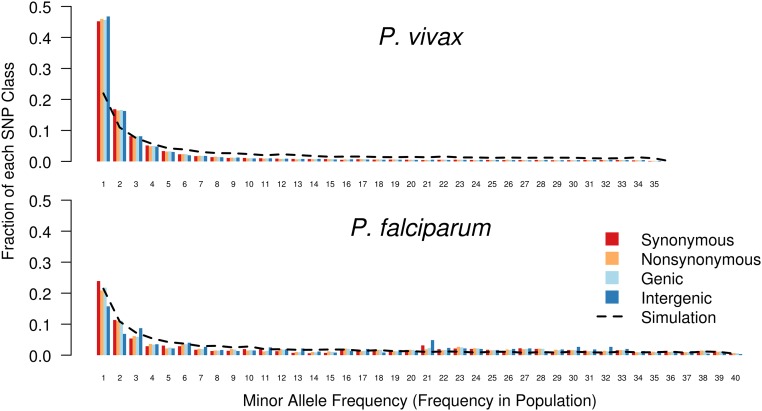
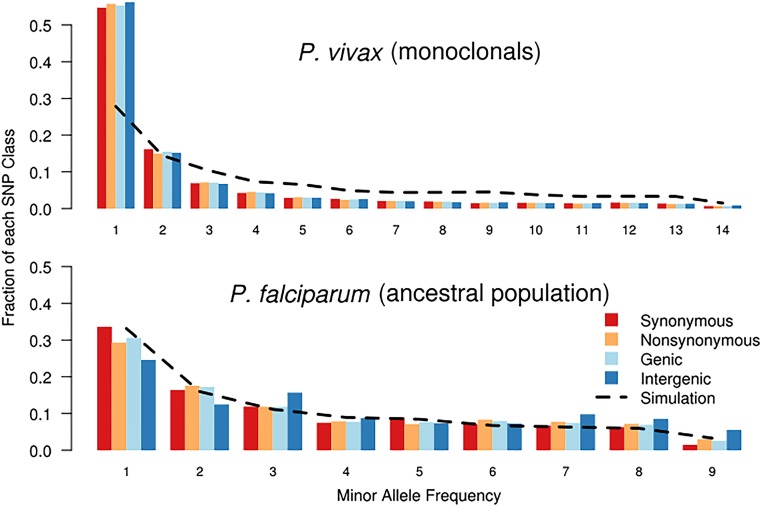
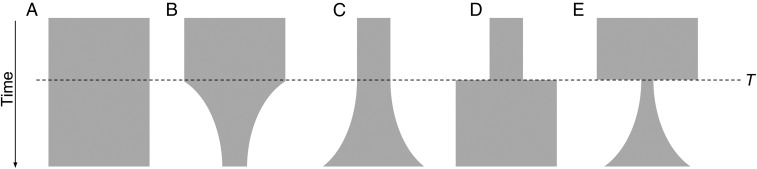
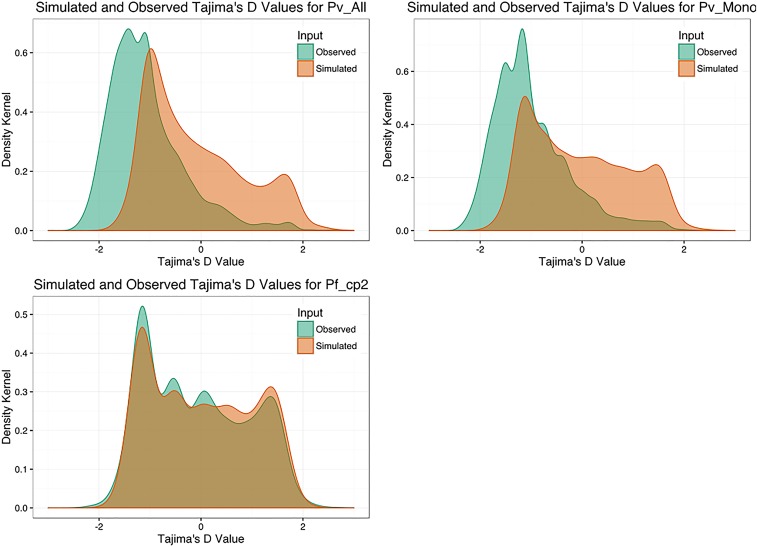
Cambodia, in which both Plasmodium vivax and Plasmodium falciparum are endemic, has been the focus of numerous malaria-control interventions, resulting in a marked decline in overall malaria incidence. Despite this decline, the number of P vivax cases has actually increased. To understand better the factors underlying this resilience, we compared the genetic responses of the two species to recent selective pressures. We sequenced and studied the genomes of 70 P vivax and 80 P falciparum isolates collected between 2009 and 2013. We found that although P falciparum has undergone population fracturing, the coendemic P vivax population has grown undisrupted, resulting in a larger effective population size, no discernable population structure, and frequent multiclonal infections. Signatures of selection suggest recent, species-specific evolutionary differences. Particularly, in contrast to P falciparum, P vivax transcription factors, chromatin modifiers, and histone deacetylases have undergone strong directional selection, including a particularly strong selective sweep at an AP2 transcription factor. Together, our findings point to different population-level adaptive mechanisms used by P vivax and P falciparum parasites. Although population substructuring in P falciparum has resulted in clonal outgrowths of resistant parasites, P vivax may use a nuanced transcriptional regulatory approach to population maintenance, enabling it to preserve a larger, more diverse population better suited to facing selective threats. We conclude that transcriptional control may underlie P vivax's resilience to malaria control measures. Novel strategies to target such processes are likely required to eradicate P vivax and achieve malaria elimination.
Keywords: Plasmodium; genome; malaria; transcription; vivax.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

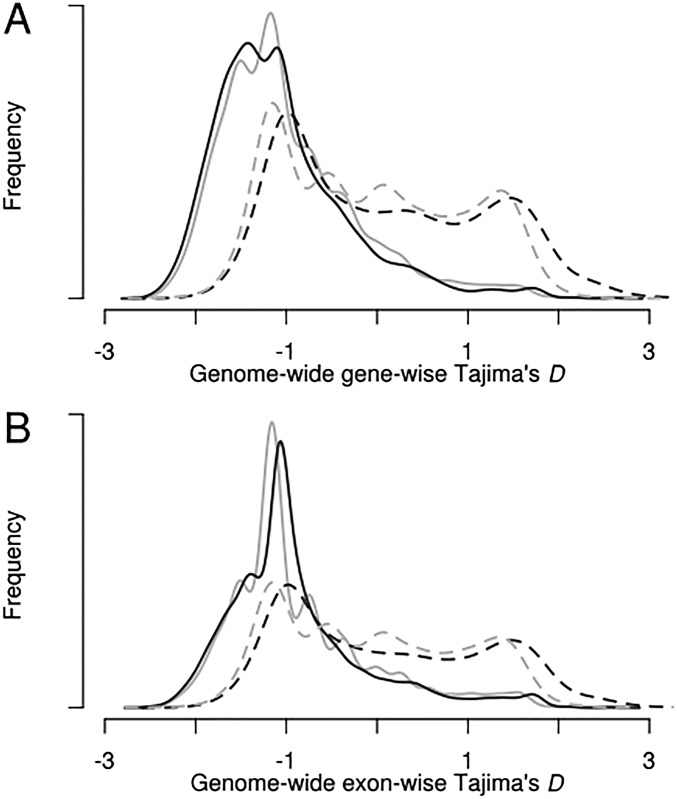
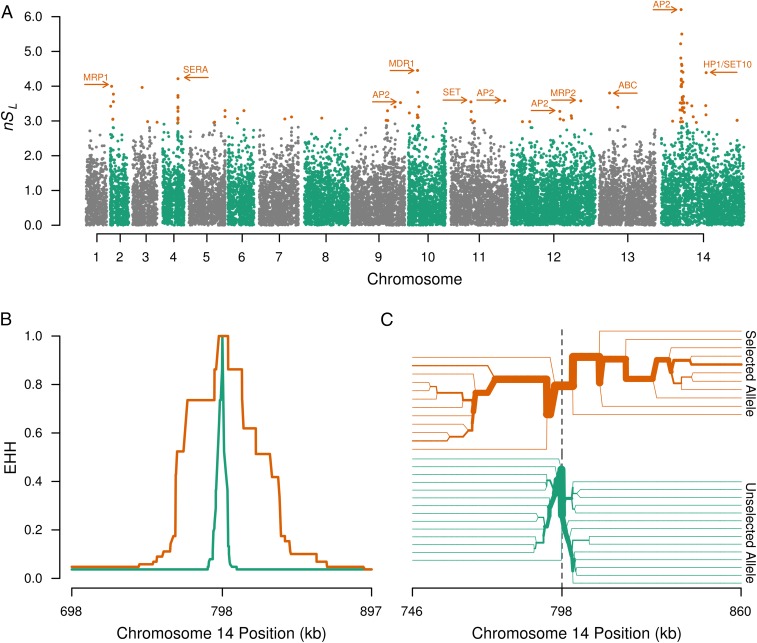
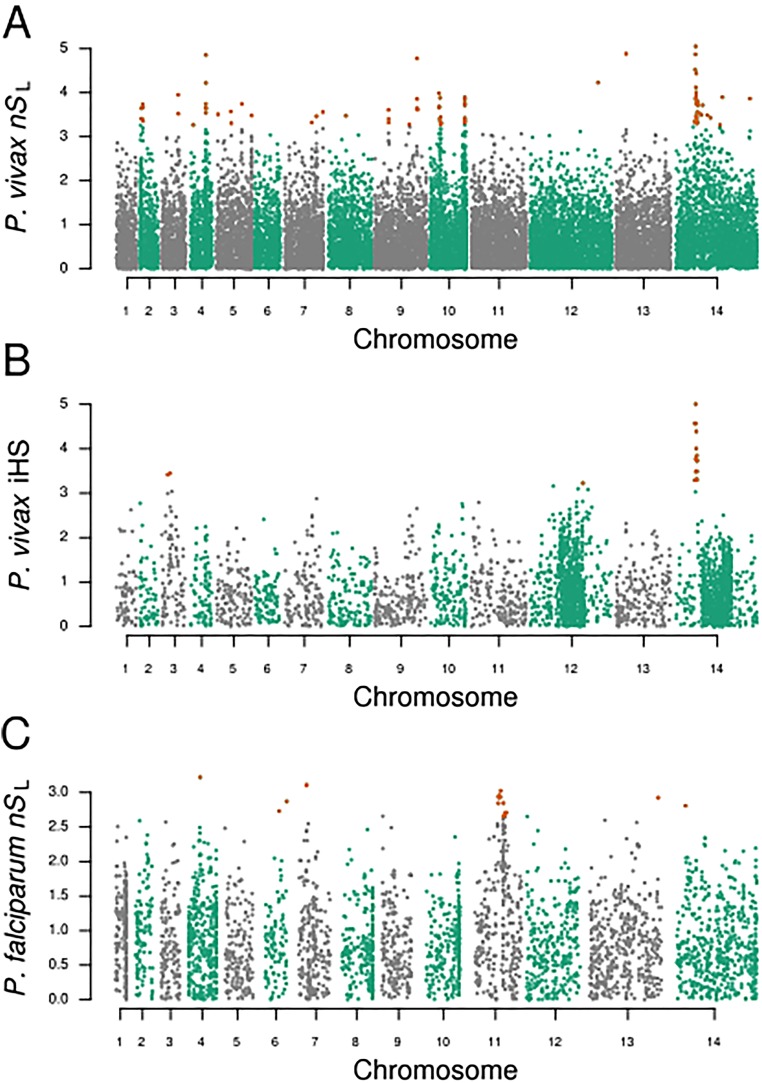
References
-
- Spring MD, et al. Dihydroartemisinin-piperaquine failure associated with a triple mutant including kelch13 C580Y in Cambodia: An observational cohort study. Lancet Infect Dis. 2015;15(6):683–691. - PubMed
-
- Zhou G, et al. Spatio-temporal distribution of Plasmodium falciparum and p. Vivax malaria in Thailand. Am J Trop Med Hyg. 2005;72(3):256–262. - PubMed
-
- Wangroongsarb P, Sudathip P, Satimai W. Characteristics and malaria prevalence of migrant populations in malaria-endemic areas along the Thai-Cambodian border. Southeast Asian J Trop Med Public Health. 2012;43(2):261–269. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
