

Mechanisms of inflammatory responses and development of insulin resistance: how are they interlinked?
- PMID: 27912756
- PMCID: PMC5135788
- DOI: 10.1186/s12929-016-0303-y
Mechanisms of inflammatory responses and development of insulin resistance: how are they interlinked?
Abstract
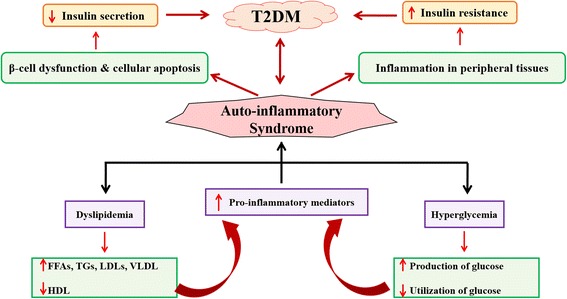
Background: Insulin resistance (IR) is one of the major hallmark for pathogenesis and etiology of type 2 diabetes mellitus (T2DM). IR is directly interlinked with various inflammatory responses which play crucial role in the development of IR. Inflammatory responses play a crucial role in the pathogenesis and development of IR which is one of the main causative factor for the etiology of T2DM.
Methods: A comprehensive online English literature was searched using various electronic search databases. Different search terms for pathogenesis of IR, role of various inflammatory responses were used and an advanced search was conducted by combining all the search fields in abstracts, keywords, and titles.
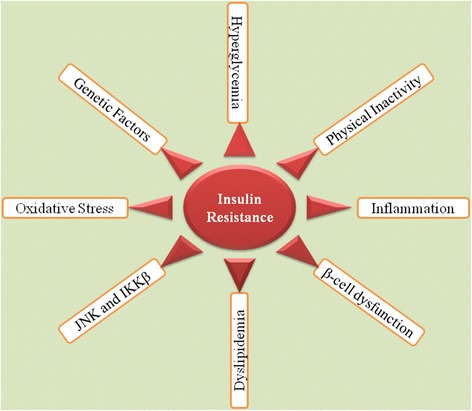
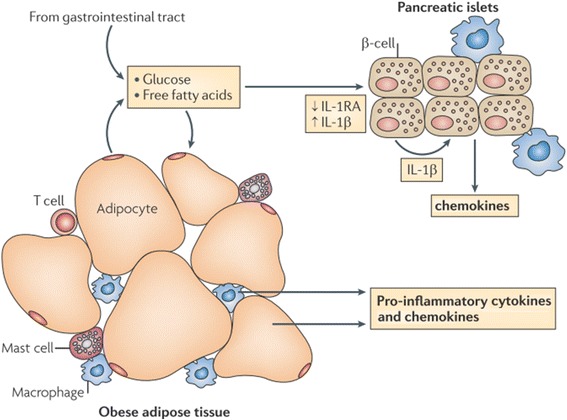
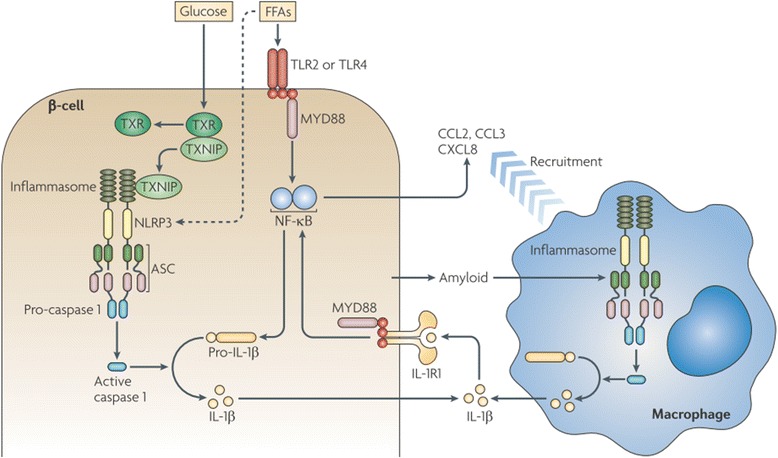
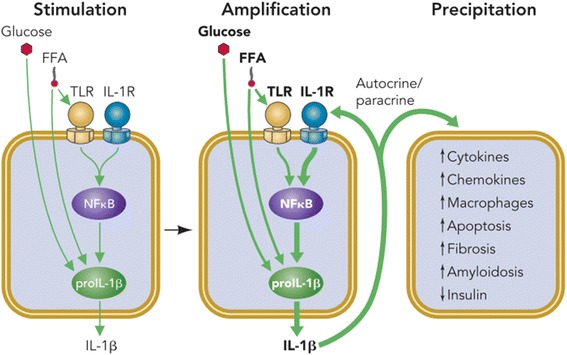
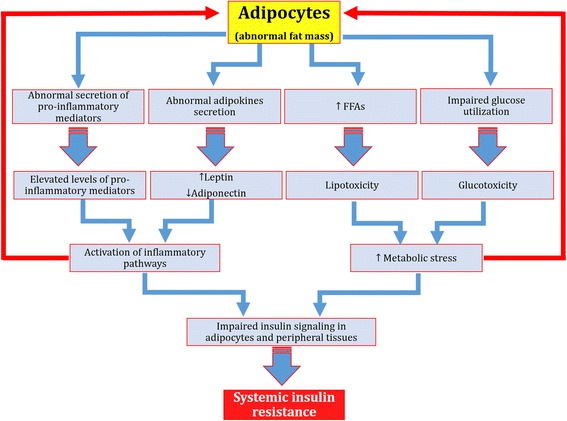
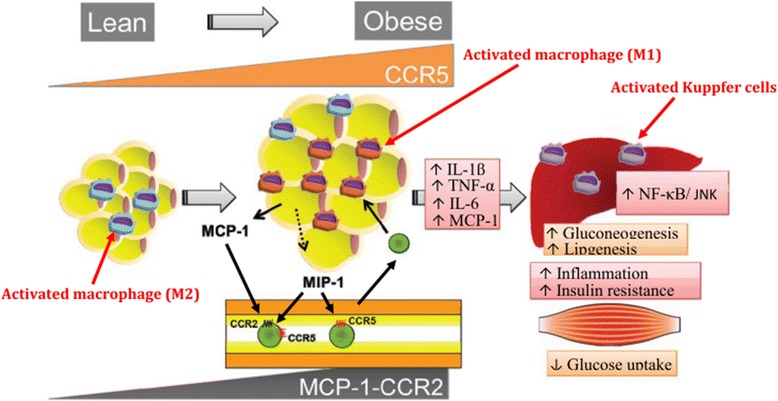
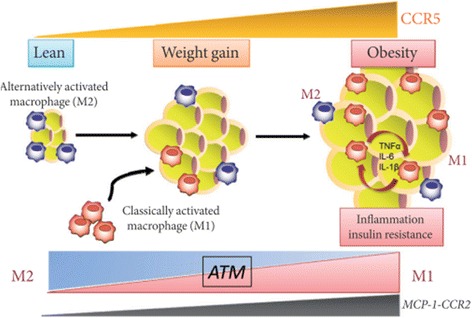
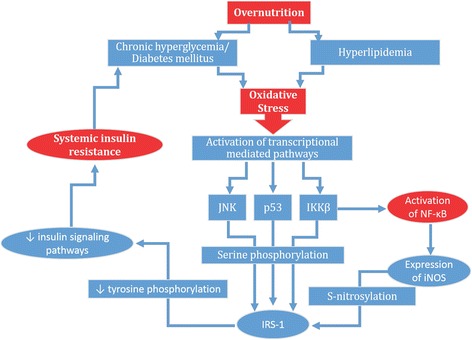
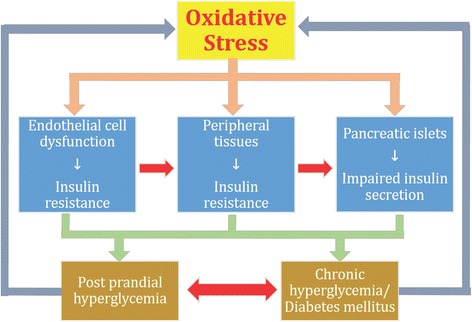
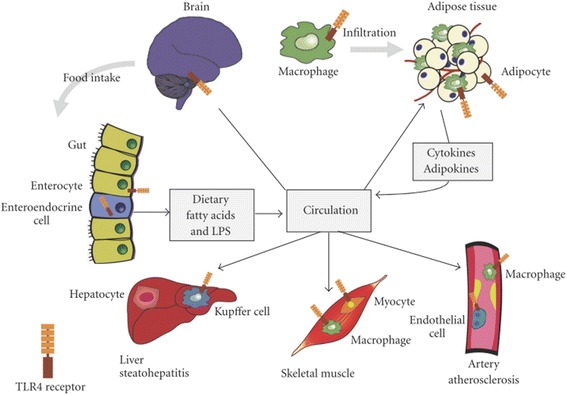
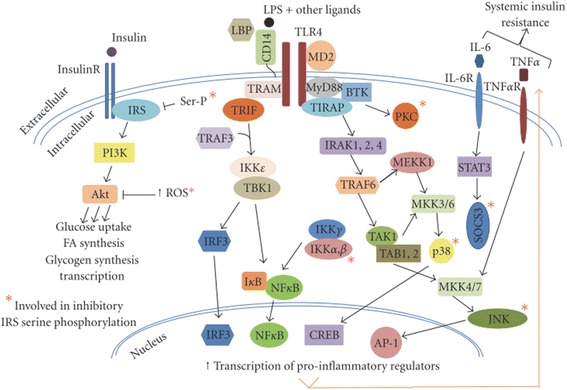
Results: We summarized the data from the searched articles and found that inflammatory responses activate the production of various pro-inflammatory mediators notably cytokines, chemokines and adipocytokines through the involvement of various transcriptional mediated molecular pathways, oxidative and metabolic stress. Overnutrition is one of the major causative factor that contributes to induce the state of low-grade inflammation due to which accumulation of elevated levels of glucose and/or lipids in blood stream occur that leads to the activation of various transcriptional mediated molecular and metabolic pathways. This results in the induction of various pro-inflammatory mediators that are decisively involved to provoke the pathogenesis of tissue-specific IR by interfering with insulin signaling pathways. Once IR is developed, it increases oxidative stress in β-cells of pancreatic islets and peripheral tissues which impairs insulin secretion, and insulin sensitivity in β-cells of pancreatic islets and peripheral tissues, respectively. Moreover, we also summarized the data regarding various treatment strategies of inflammatory responses-induced IR.
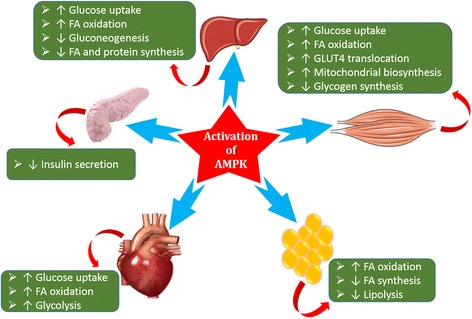
Conclusions: In this article, we have briefly described that how pro-inflammatory mediators, oxidative stress, transcriptional mediated molecular and metabolic pathways are involved in the pathogenesis of tissues-specific IR. Moreover, based on recent investigations, we have also described that to counterfeit these inflammatory responses is one of the best treatment strategy to prevent the pathogenesis of IR through ameliorating the incidences of inflammatory responses.
Keywords: Diabetes mellitus; Insulin resistance; Insulin sensitivity; Pro-inflammatory mediators; Transcriptional pathways.
Figures
References
-
- Rehman K. and Akash M.S.H. Nutrition and Diabetes Mellitus: How are They Interlinked? 2016;26(4):317–332. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
