

Atypical hemolytic uremic syndrome
- PMID: 27913483
- PMCID: PMC6142509
- DOI: 10.1182/asheducation-2016.1.217
Atypical hemolytic uremic syndrome
Abstract
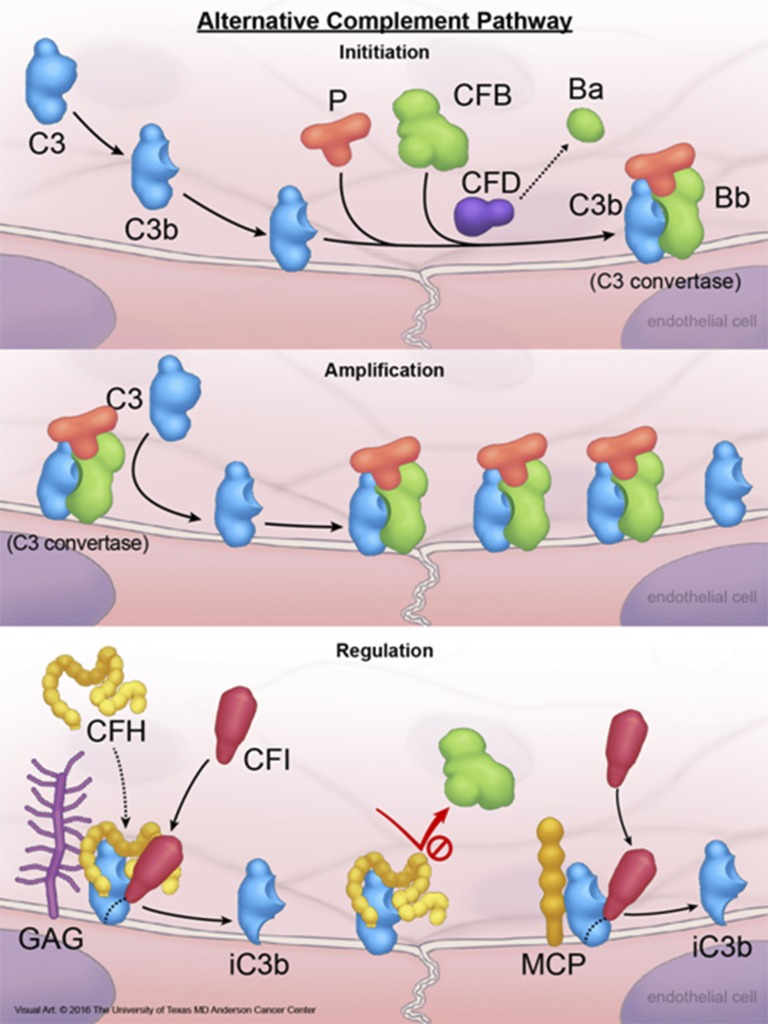
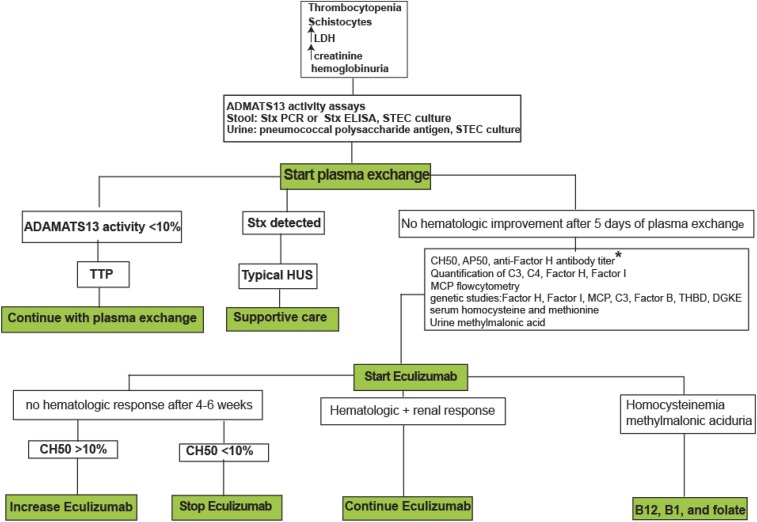
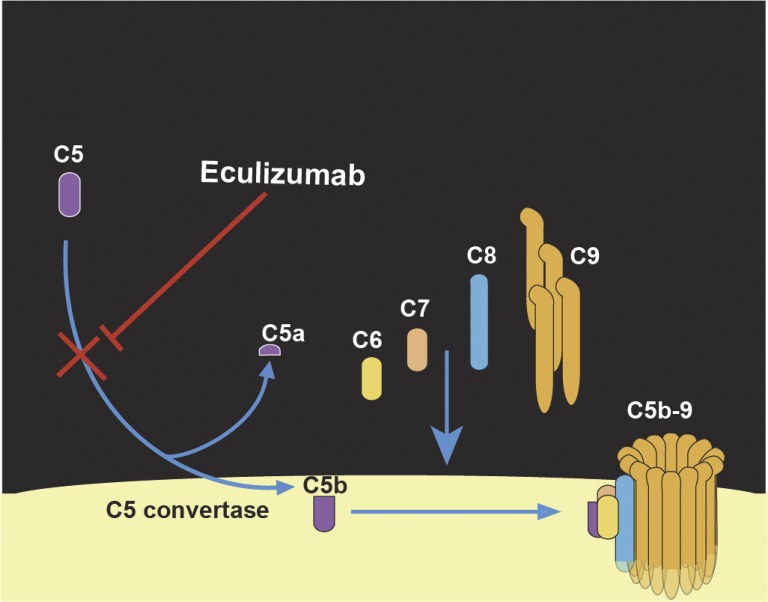
Atypical hemolytic uremic syndrome (aHUS) is a thrombotic microangiopathy (TMA) that affects multiple organs and the kidneys in particular. aHUS can be sporadic or familial and is most commonly caused by dysregulation of the alternative complement pathway. The initial attack of aHUS can occur at any age, and is associated with a high rate of progression to end stage renal disease. Many aHUS patients relapse in the native or transplanted kidneys, and require close monitoring and long-term management. Availability of anticomplement therapy has revolutionized the management of aHUS, and can change the natural course of aHUS by inducing hematologic remission, improving or stabilizing kidney functions, and preventing graft failure. As a result, it is important to succeed in the challenging task of differentiating aHUS from other TMAs and initiate adequate treatment early during the course of disease. Considering the high cost of currently available anticomplement therapy, it is important also from a financial point of view to accurately diagnose aHUS early during the course of disease and determine the necessary length of therapy. This highlights the need for development of precise complement functional and genetic studies with rapid turnaround time.
© 2016 by The American Society of Hematology. All rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: V.A.-K. is on the Board of Directors or on an advisory committee for Alexion.
Figures
References
-
- George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371(7):654-666. - PubMed
-
- Moake JL. Thrombotic microangiopathies. N Engl J Med. 2002;347(8):589-600. - PubMed
-
- Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365(9464):1073-1086. - PubMed
-
- Schwartz J, Winters JL, Padmanabhan A, et al. . Guidelines on the use of therapeutic apheresis in clinical practice-evidence-based approach from the Writing Committee of the American Society for Apheresis: the sixth special issue. J Clin Apher. 2013;28(3):145-284. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials