

Sickle cell disease in the older adult
- PMID: 27914684
- PMCID: PMC10757825
- DOI: 10.1016/j.pathol.2016.10.002
Sickle cell disease in the older adult
Abstract
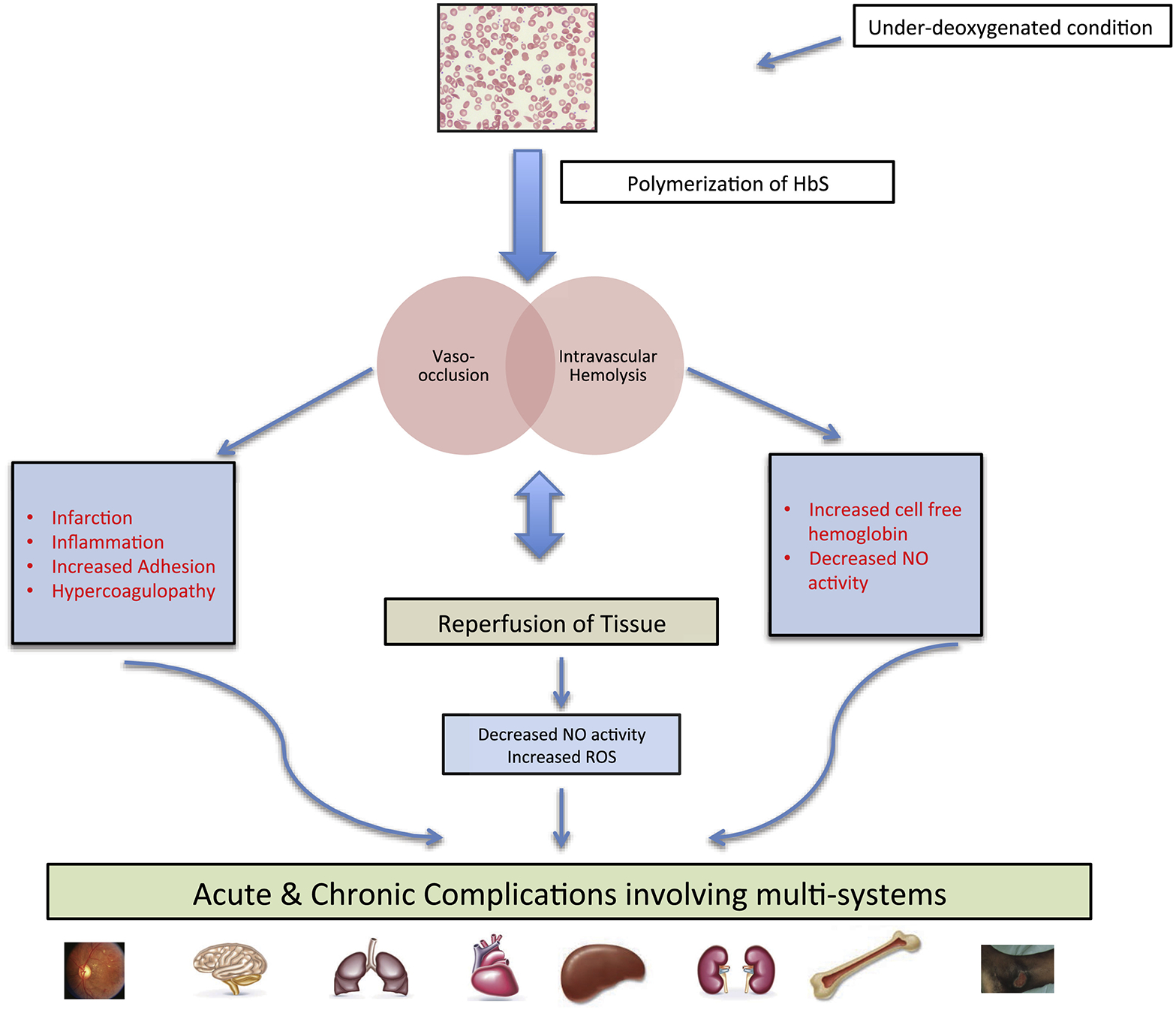
Sickle cell disease (SCD) is an inherited haemoglobin disorder, associated with recurrent painful episodes, ongoing haemolytic anaemia and progressive multi-organ damage. Until the early 1990s, survival beyond the fourth decade for a patient with SCD was considered unusual and prompted case reports. Nowadays, in countries with developed health care systems, more than 90 percent of newborns with SCD survive into adulthood. Nevertheless, their life expectancy is still shortened by more than two decades compared to the general population. With an increasing life expectancy, SCD has now evolved into a debilitating disorder with substantial morbidity resulting from ongoing sickle cell vasculopathy and multi-organ damage. Limited data on health care issues of older adults with SCD poses multiple challenges to patients, their families and health care providers. In this review, we will address and discuss acute and chronic complications of SCD with a special focus on the older adult.
Keywords: Sickle cell disease; multi-organ damage; older adults; vasculopathy.
Published by Elsevier B.V.
Conflict of interest statement
Figures


References
-
- Aygun B, Odame I. A global perspective on sickle cell disease. Pediatr Blood Cancer 2012; 59: 386–90. - PubMed
-
- Hamideh D, Alvarez O. Sickle cell disease related mortality in the United States (1999–2009). Pediatr Blood Cancer 2013; 60: 1482–6. - PubMed
-
- Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994; 330: 1639–44. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
