

Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism
- PMID: 27919067
- PMCID: PMC7102905
- DOI: 10.1038/nature20612
Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism
Erratum in
-
Author Correction: Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism.Nature. 2018 Aug;560(7718):E30. doi: 10.1038/s41586-018-0295-8. Nature. 2018. PMID: 29995847
Abstract
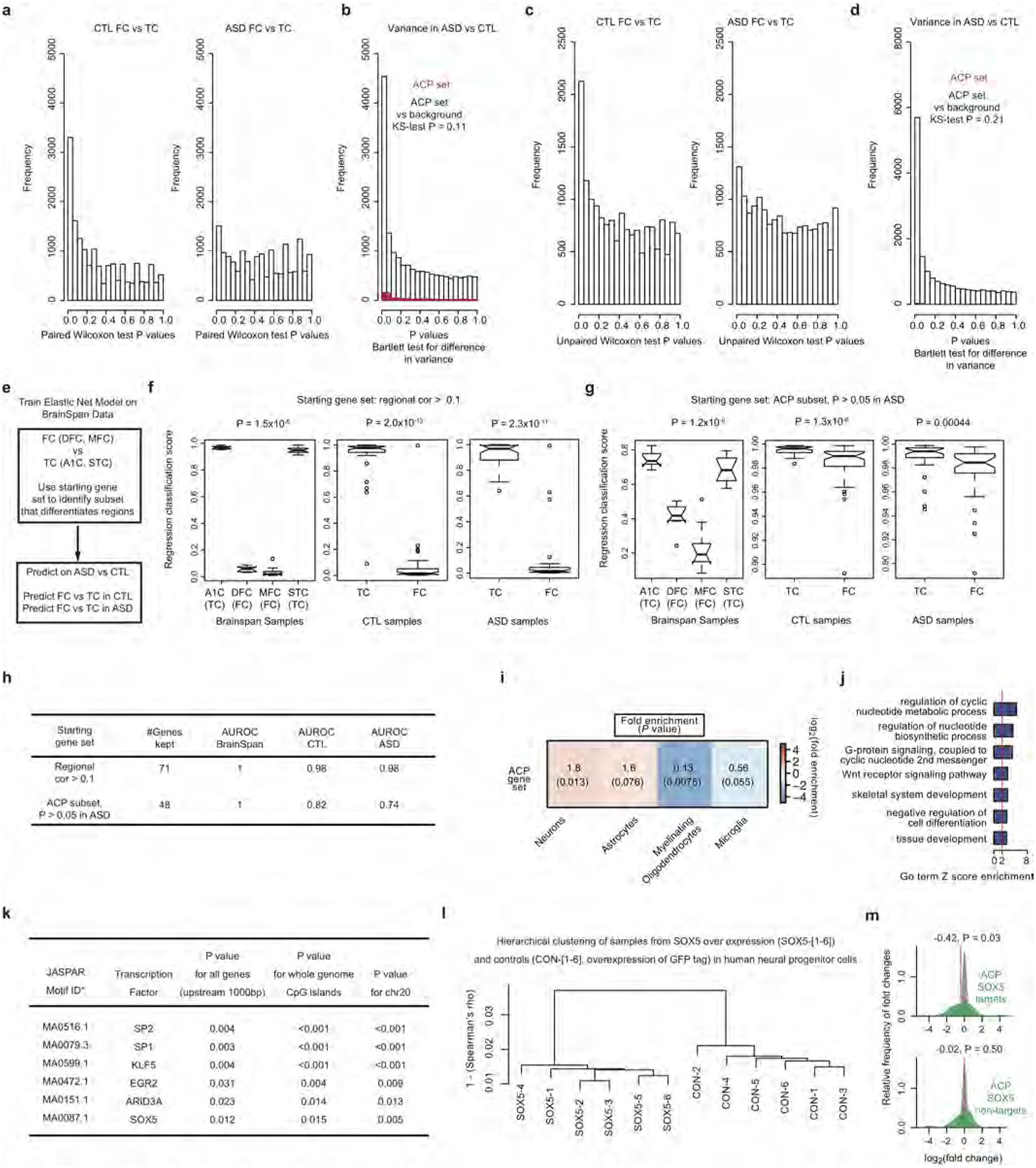
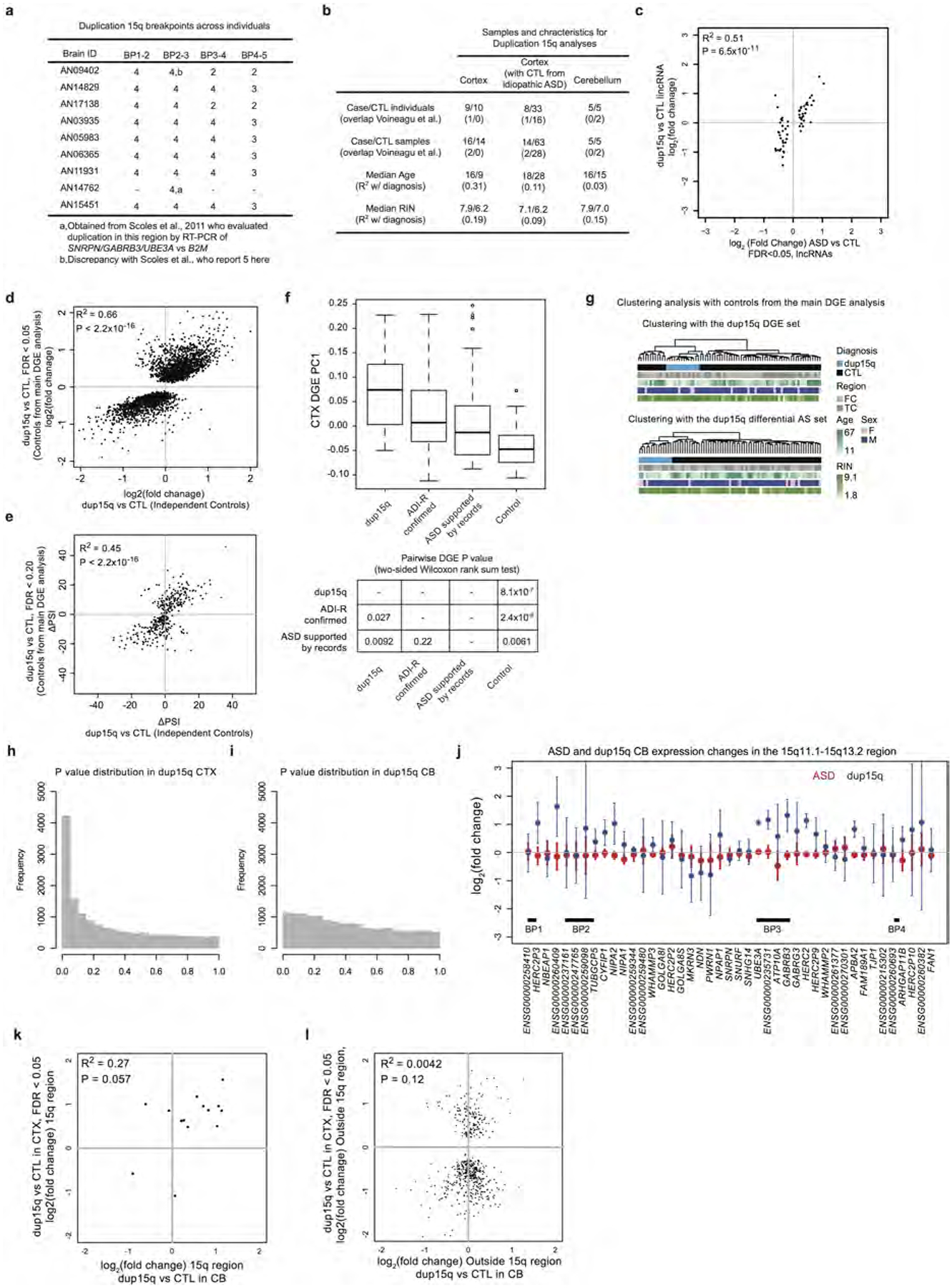
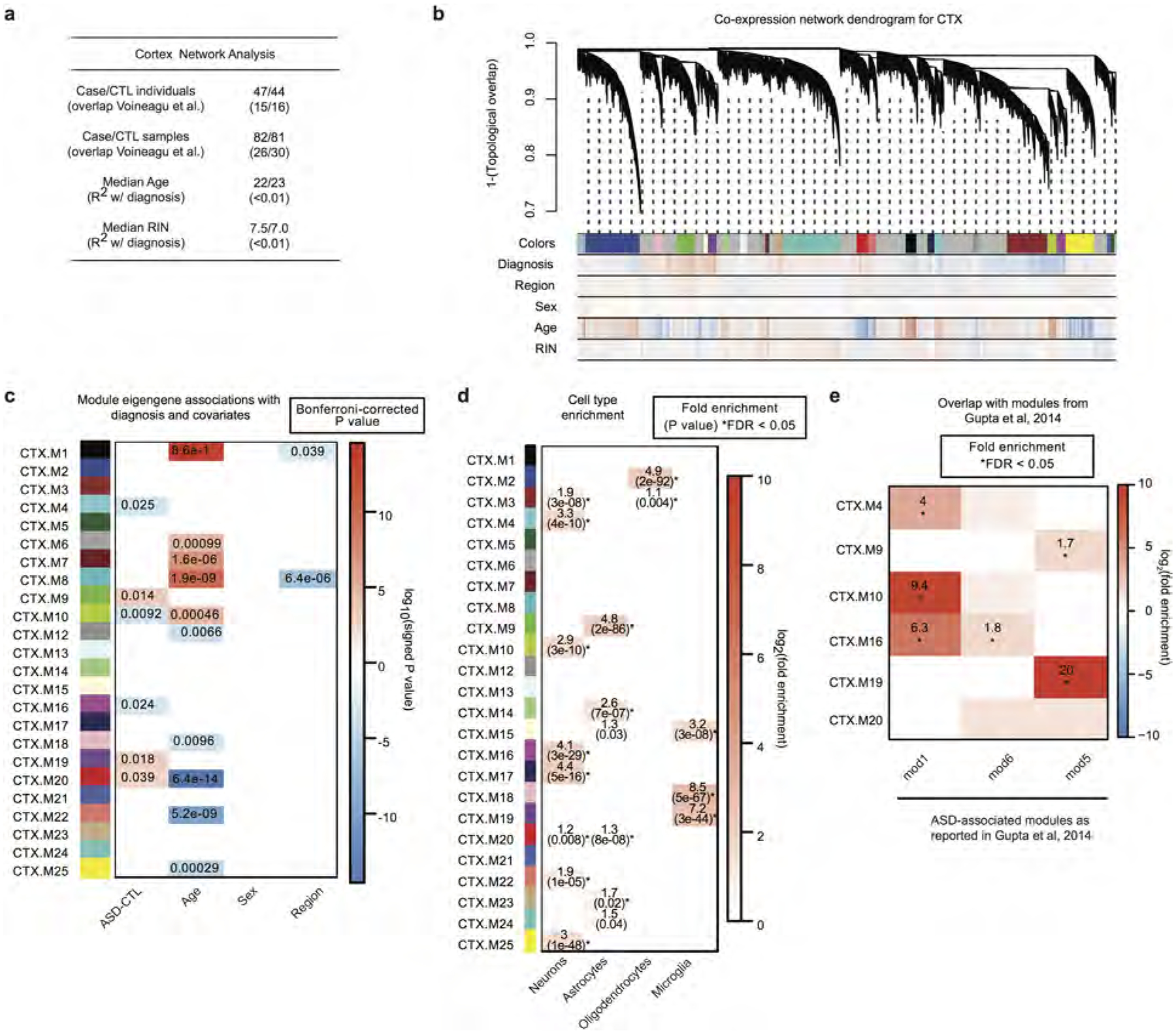
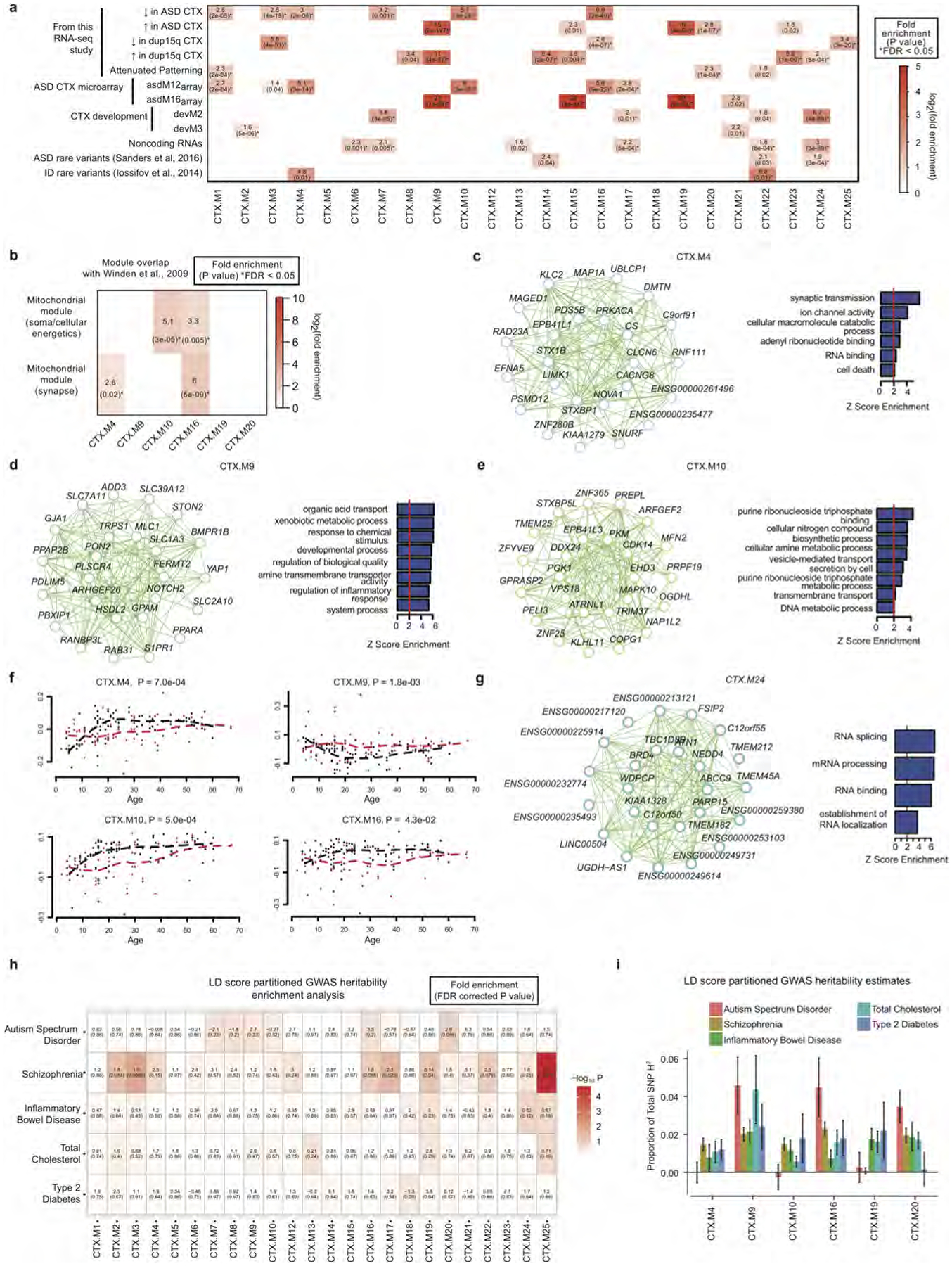
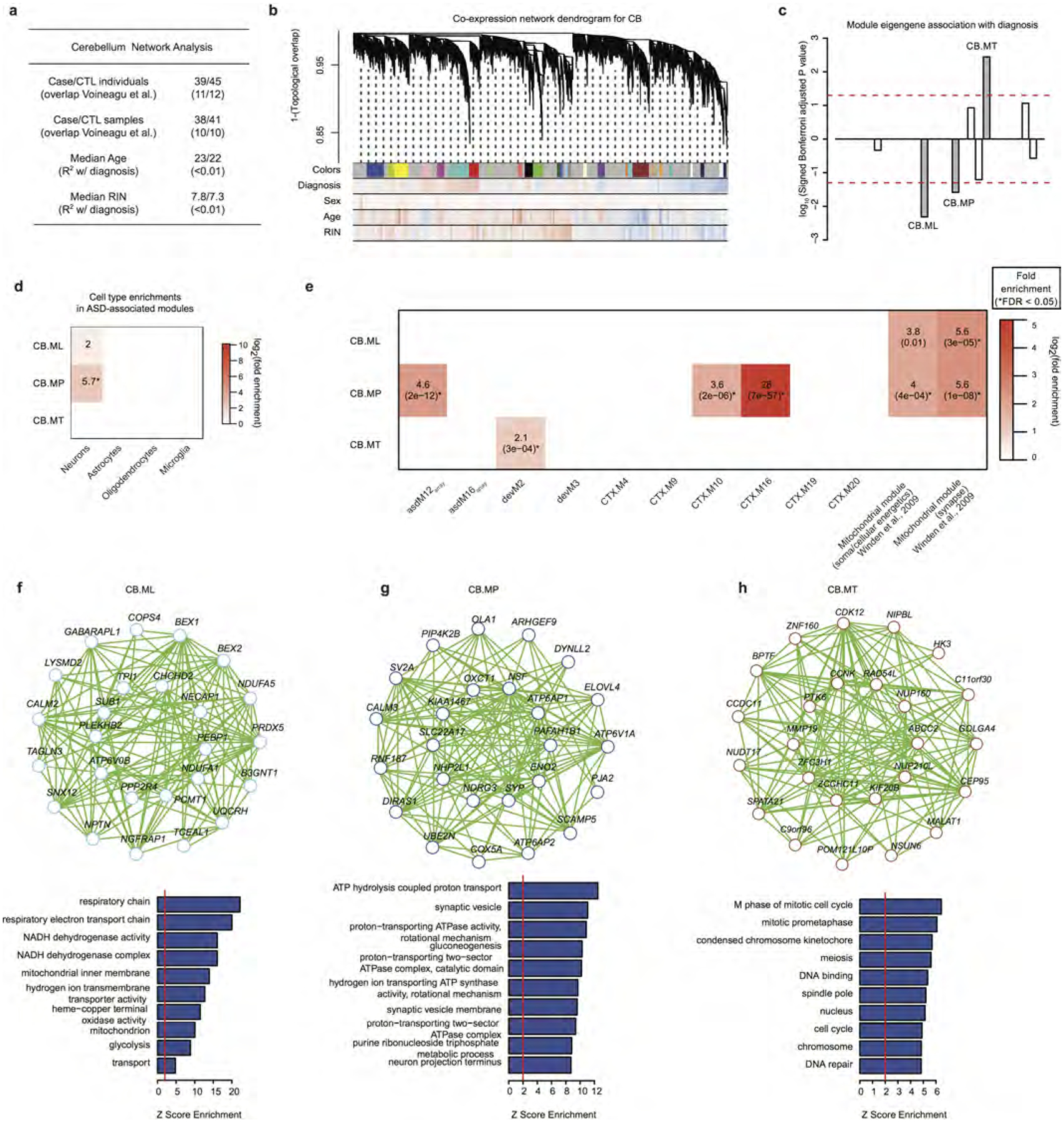
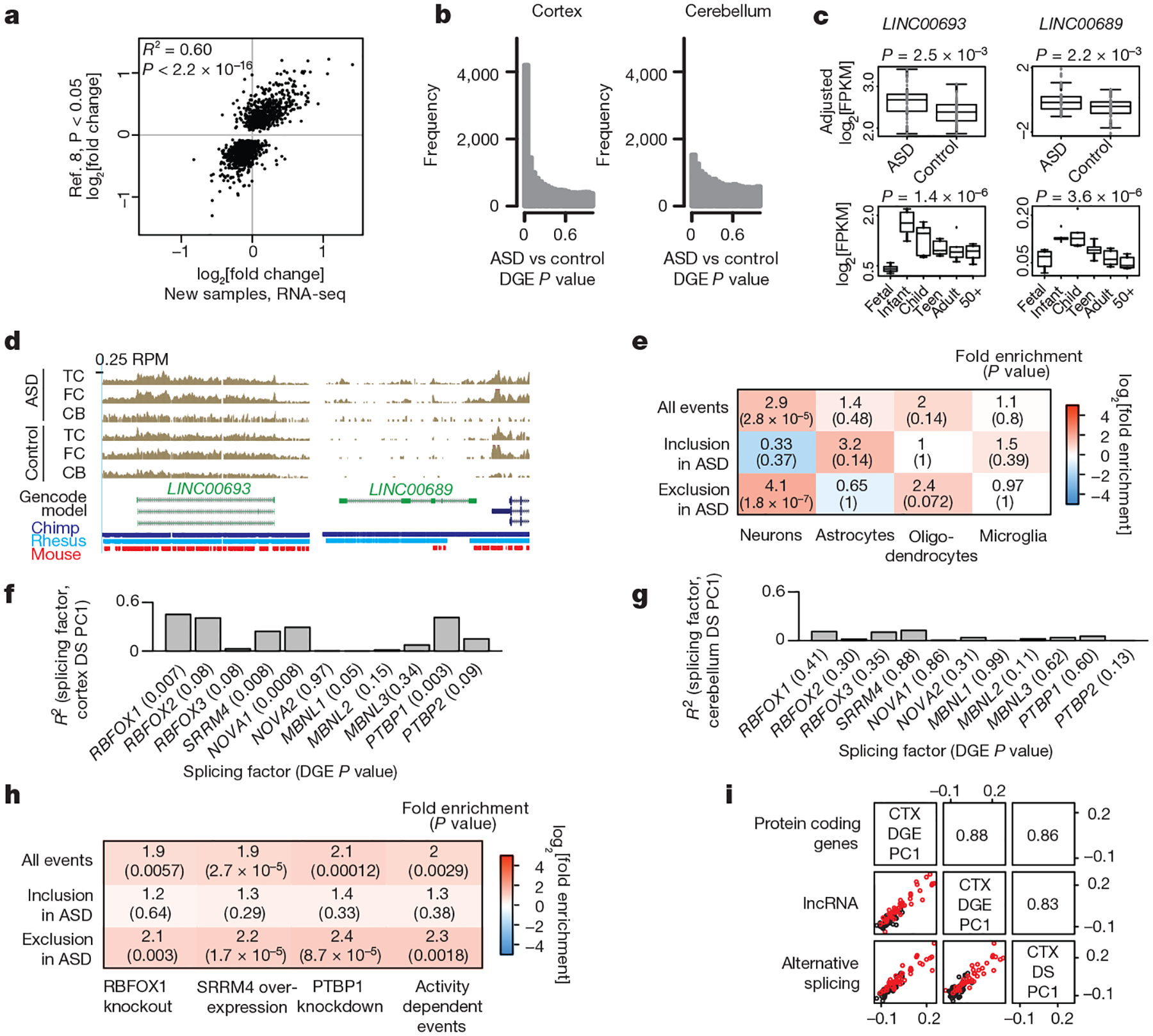
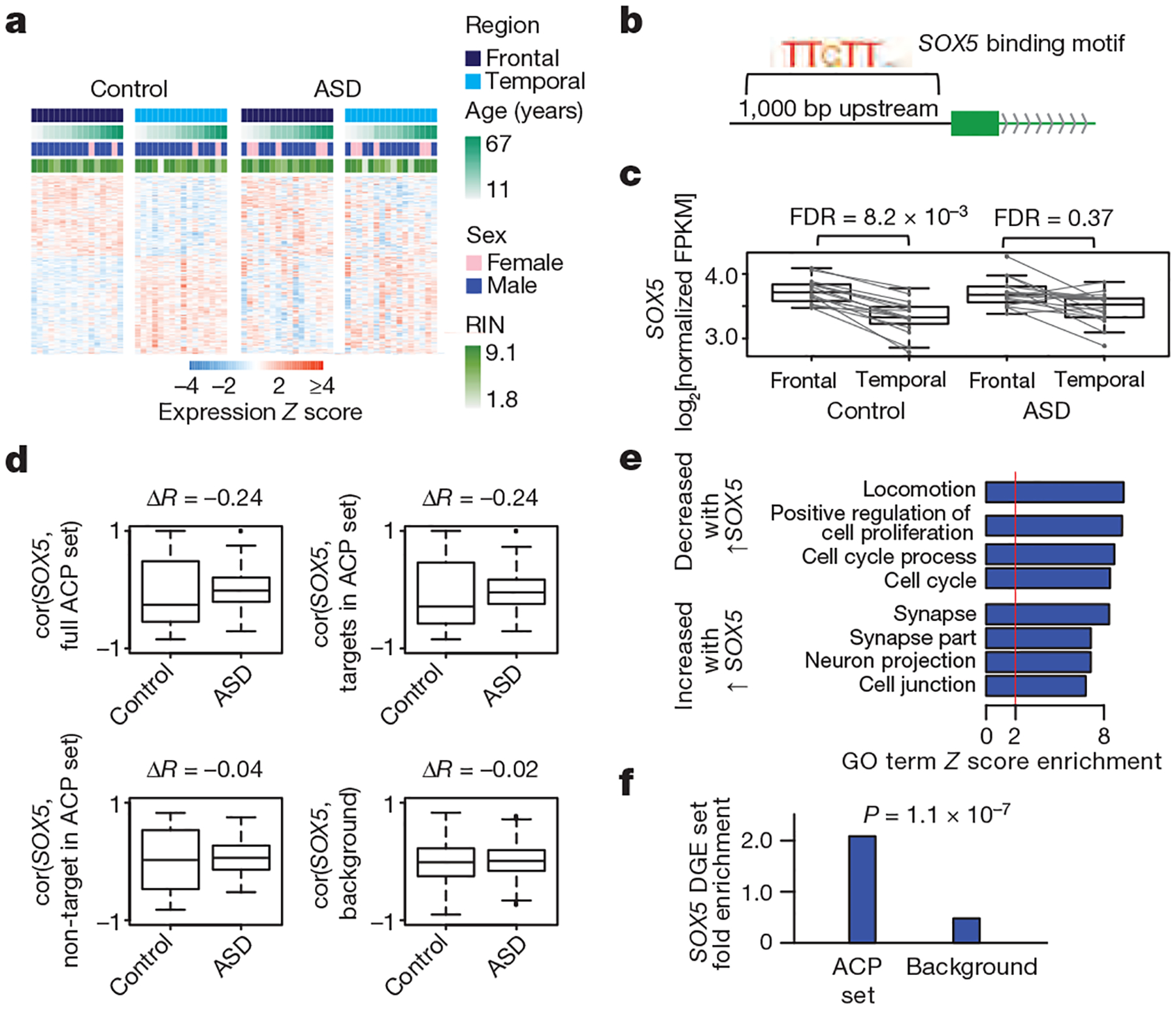
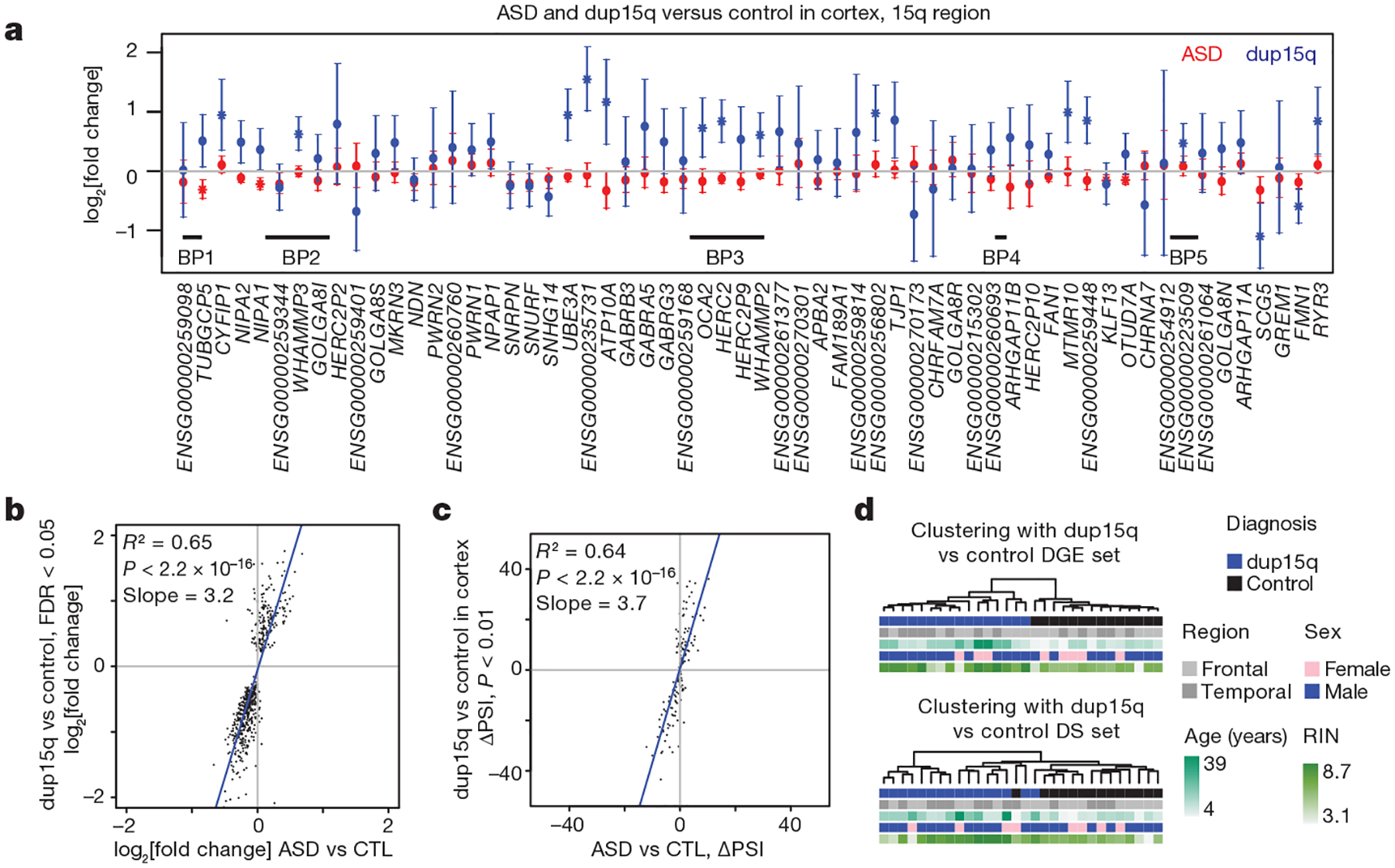
Autism spectrum disorder (ASD) involves substantial genetic contributions. These contributions are profoundly heterogeneous but may converge on common pathways that are not yet well understood. Here, through post-mortem genome-wide transcriptome analysis of the largest cohort of samples analysed so far, to our knowledge, we interrogate the noncoding transcriptome, alternative splicing, and upstream molecular regulators to broaden our understanding of molecular convergence in ASD. Our analysis reveals ASD-associated dysregulation of primate-specific long noncoding RNAs (lncRNAs), downregulation of the alternative splicing of activity-dependent neuron-specific exons, and attenuation of normal differences in gene expression between the frontal and temporal lobes. Our data suggest that SOX5, a transcription factor involved in neuron fate specification, contributes to this reduction in regional differences. We further demonstrate that a genetically defined subtype of ASD, chromosome 15q11.2-13.1 duplication syndrome (dup15q), shares the core transcriptomic signature observed in idiopathic ASD. Co-expression network analysis reveals that individuals with ASD show age-related changes in the trajectory of microglial and synaptic function over the first two decades, and suggests that genetic risk for ASD may influence changes in regional cortical gene expression. Our findings illustrate how diverse genetic perturbations can lead to phenotypic convergence at multiple biological levels in a complex neuropsychiatric disorder.
Conflict of interest statement
The authors declare competing financial interests: details are available in the online version of the paper. Readers are welcome to comment on the online version of the paper.
Figures
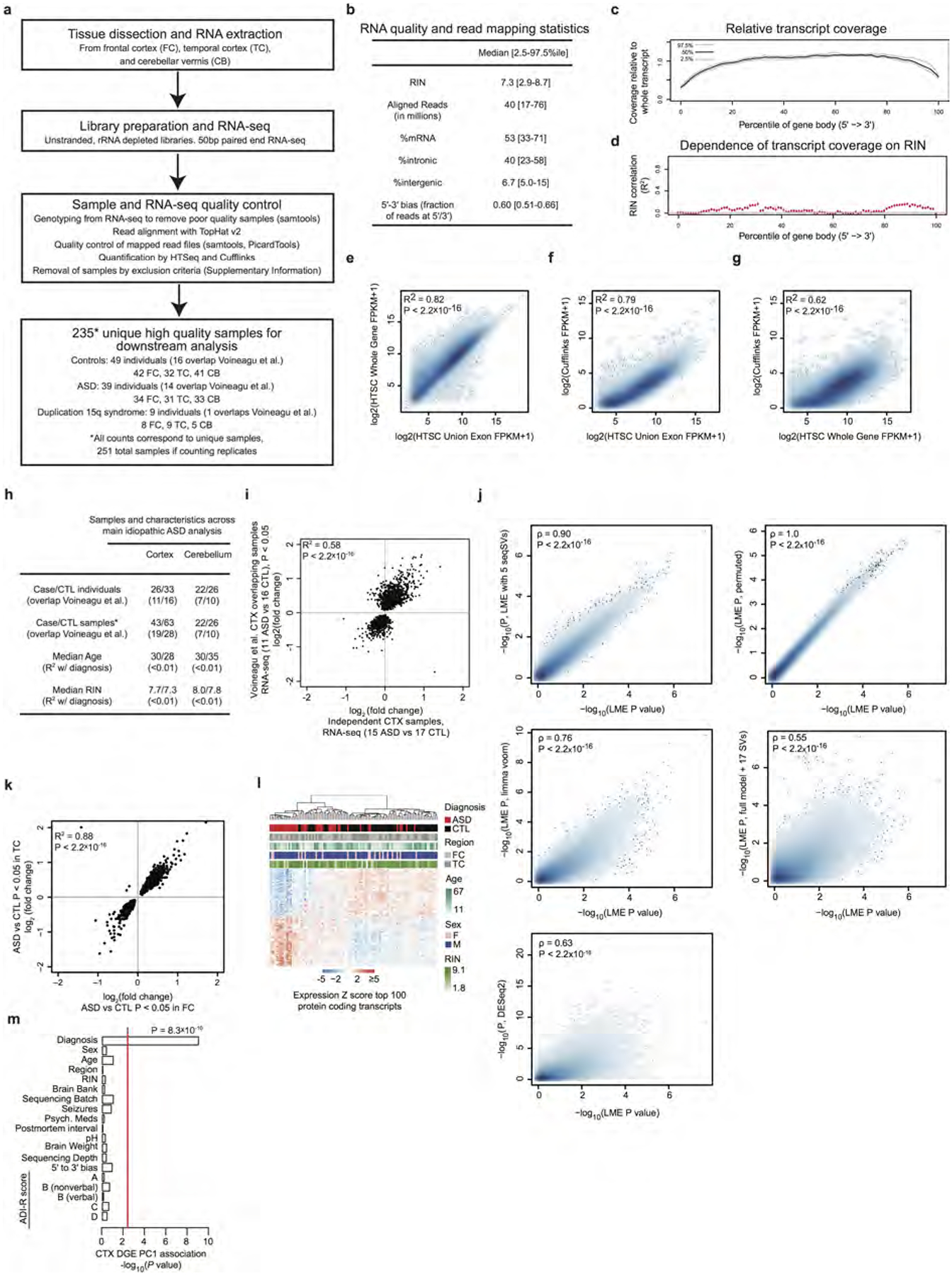
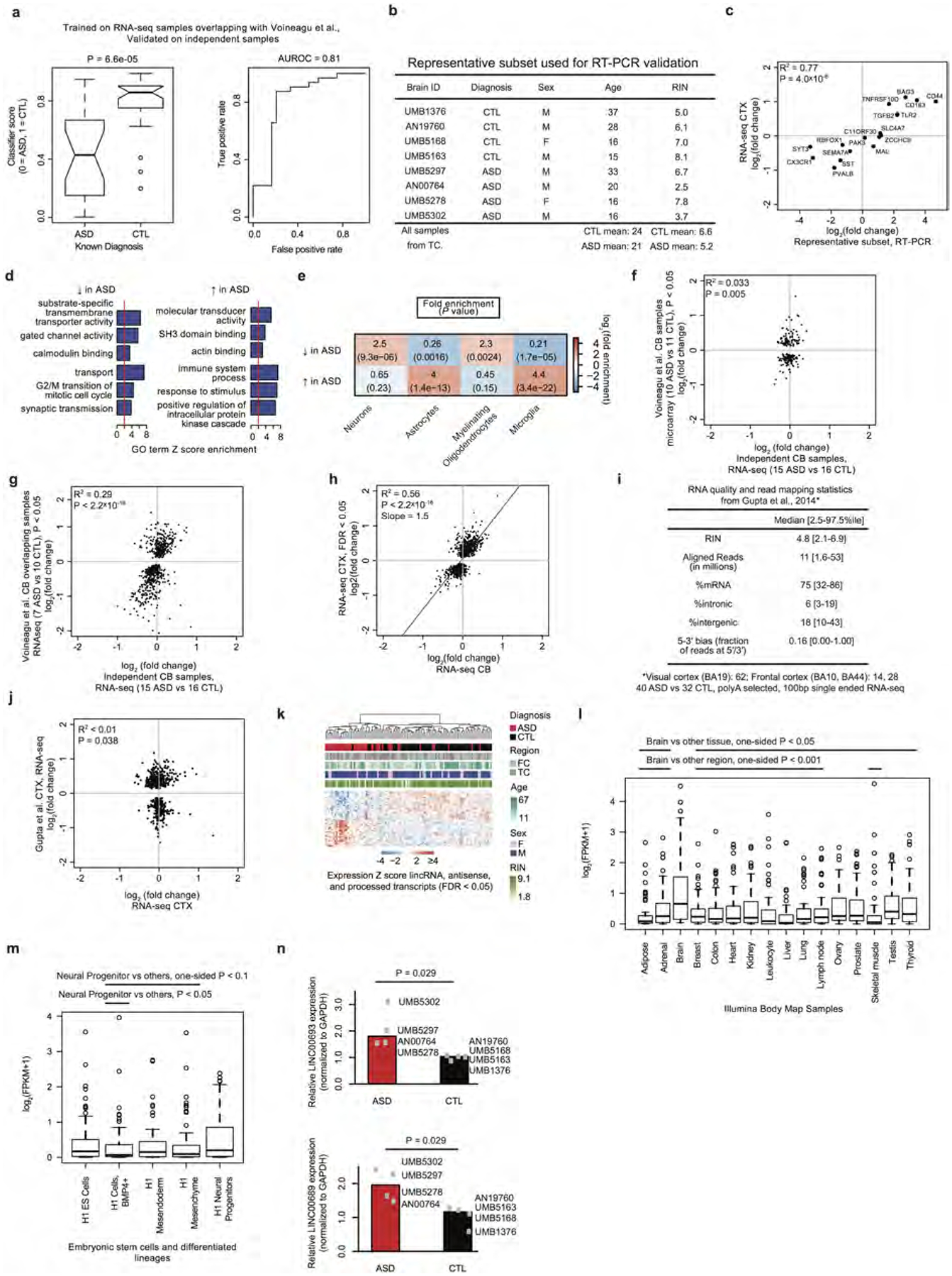

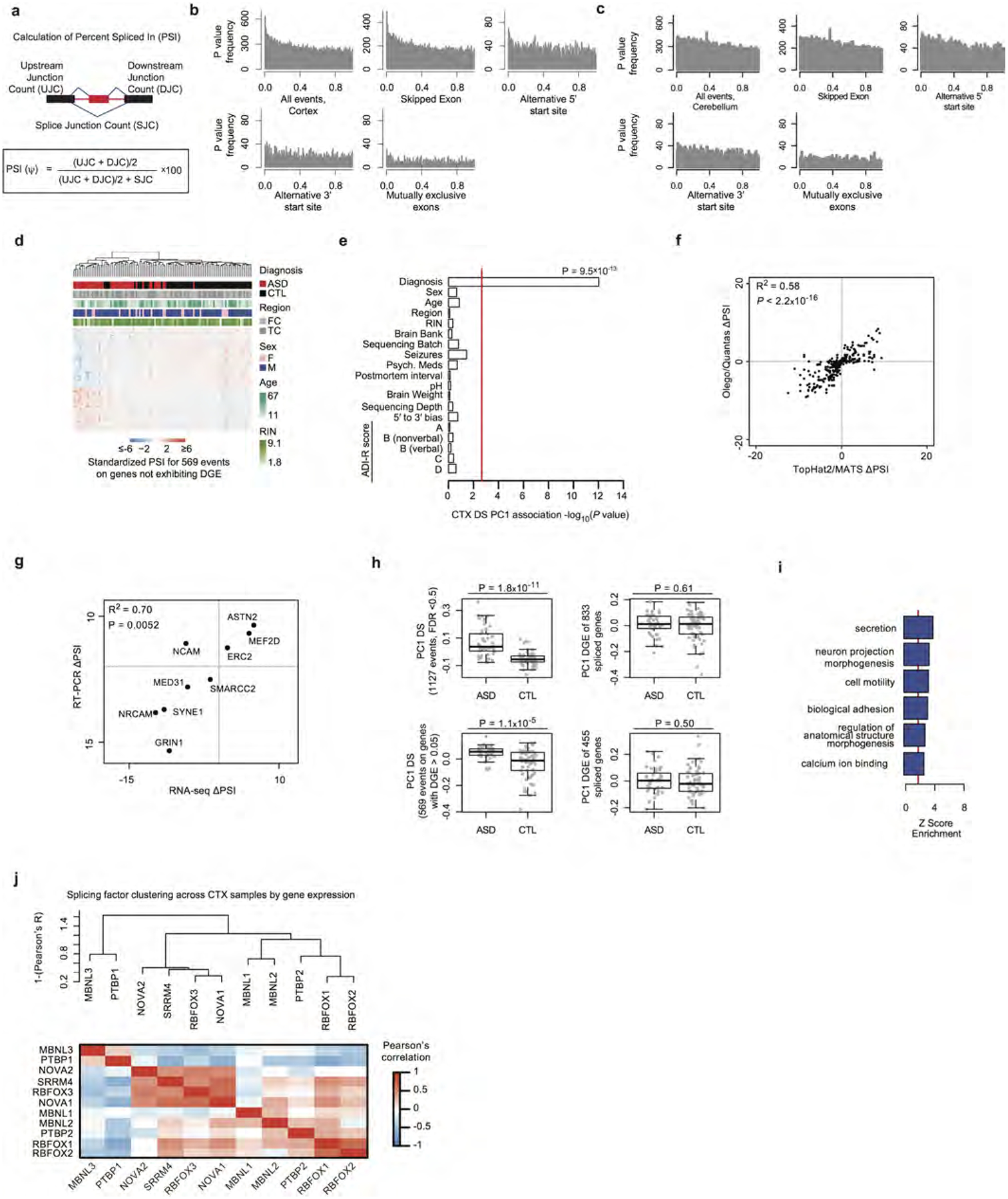
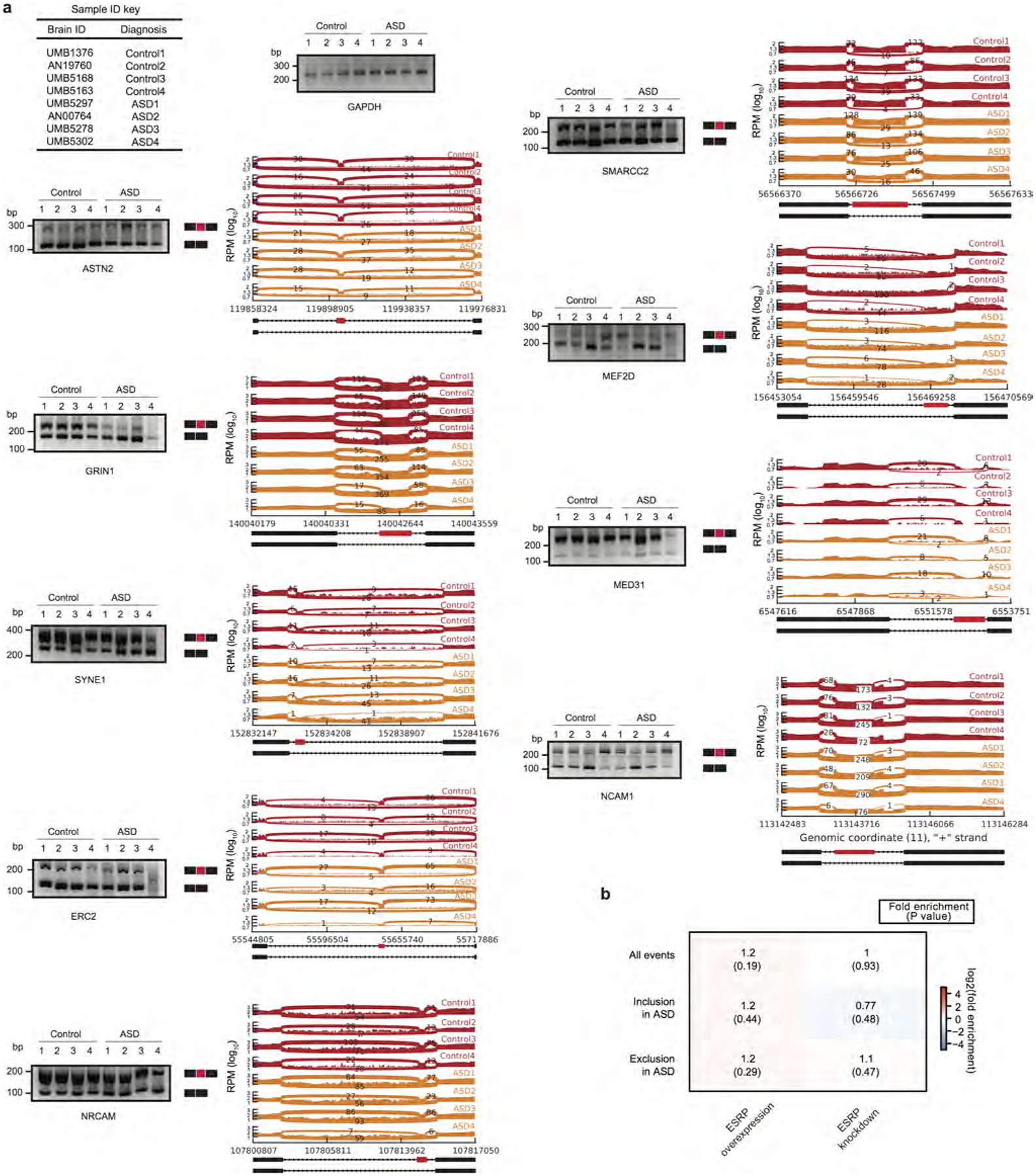
Comment in
-
Neurodevelopmental disorders: Converging on autism spectrum disorder.Nat Rev Neurosci. 2017 Feb;18(2):67. doi: 10.1038/nrn.2016.184. Epub 2017 Jan 5. Nat Rev Neurosci. 2017. PMID: 28053329 No abstract available.
References
-
- Gratten J, Visscher PM, Mowry BJ & Wray NR Interpreting the role of de novo protein-coding mutations in neuropsychiatric disease. Nat. Genet 45, 234–238 (2013). - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
