

MEK inhibitors block growth of lung tumours with mutations in ataxia-telangiectasia mutated
- PMID: 27922010
- PMCID: PMC5150652
- DOI: 10.1038/ncomms13701
MEK inhibitors block growth of lung tumours with mutations in ataxia-telangiectasia mutated
Abstract
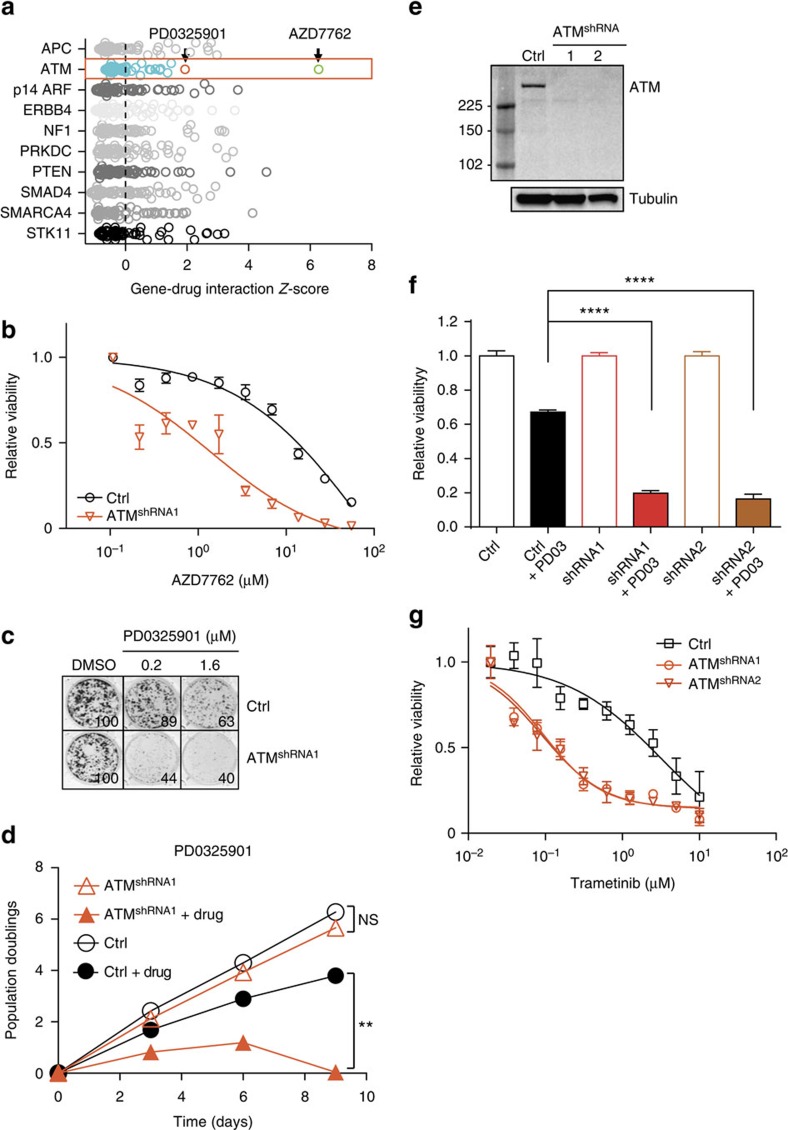
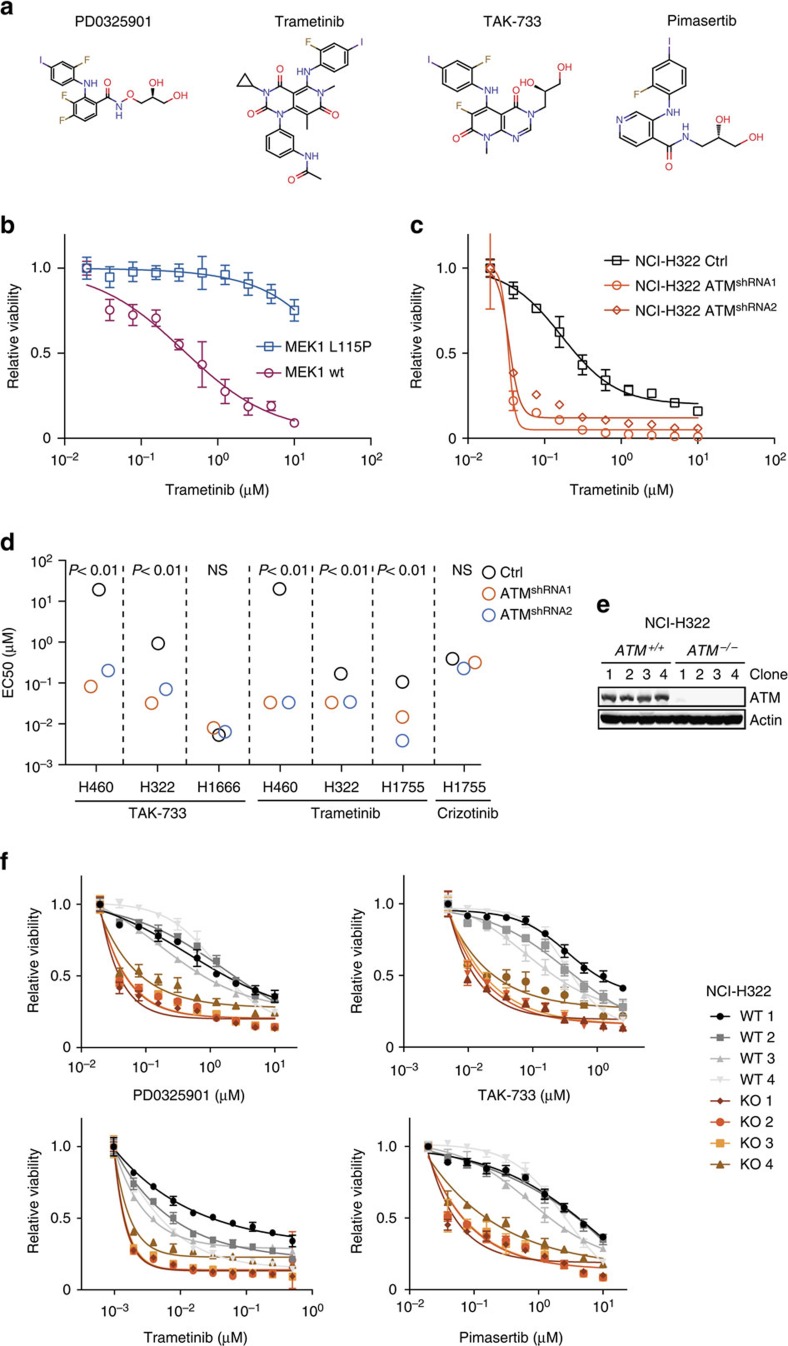
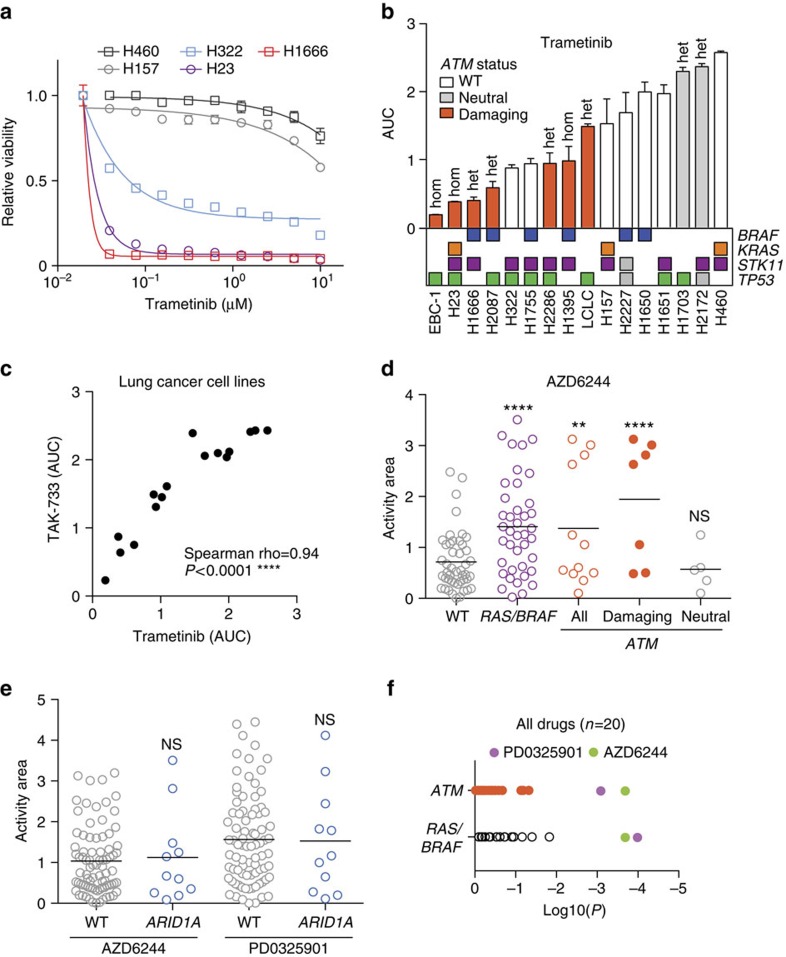
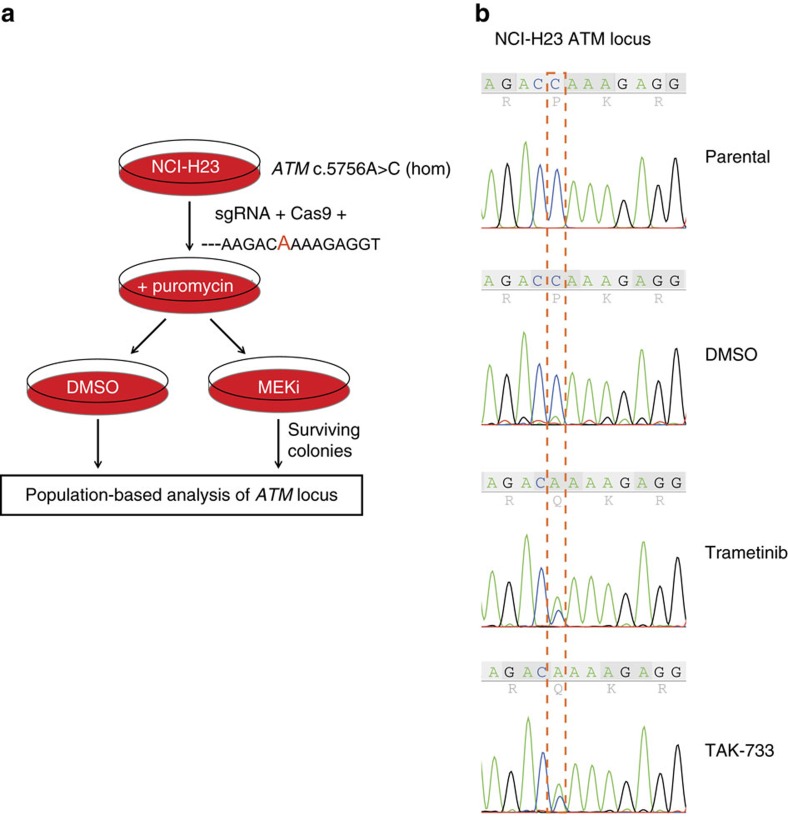
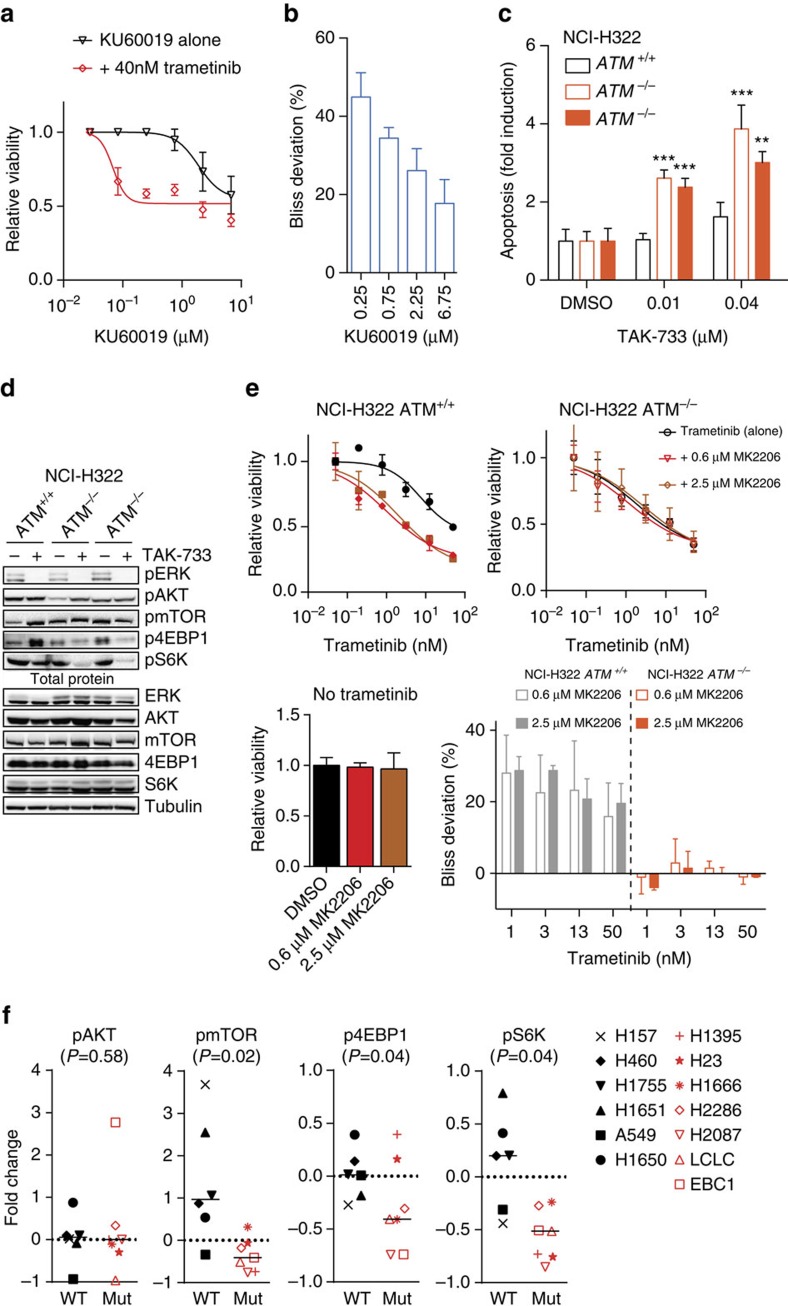
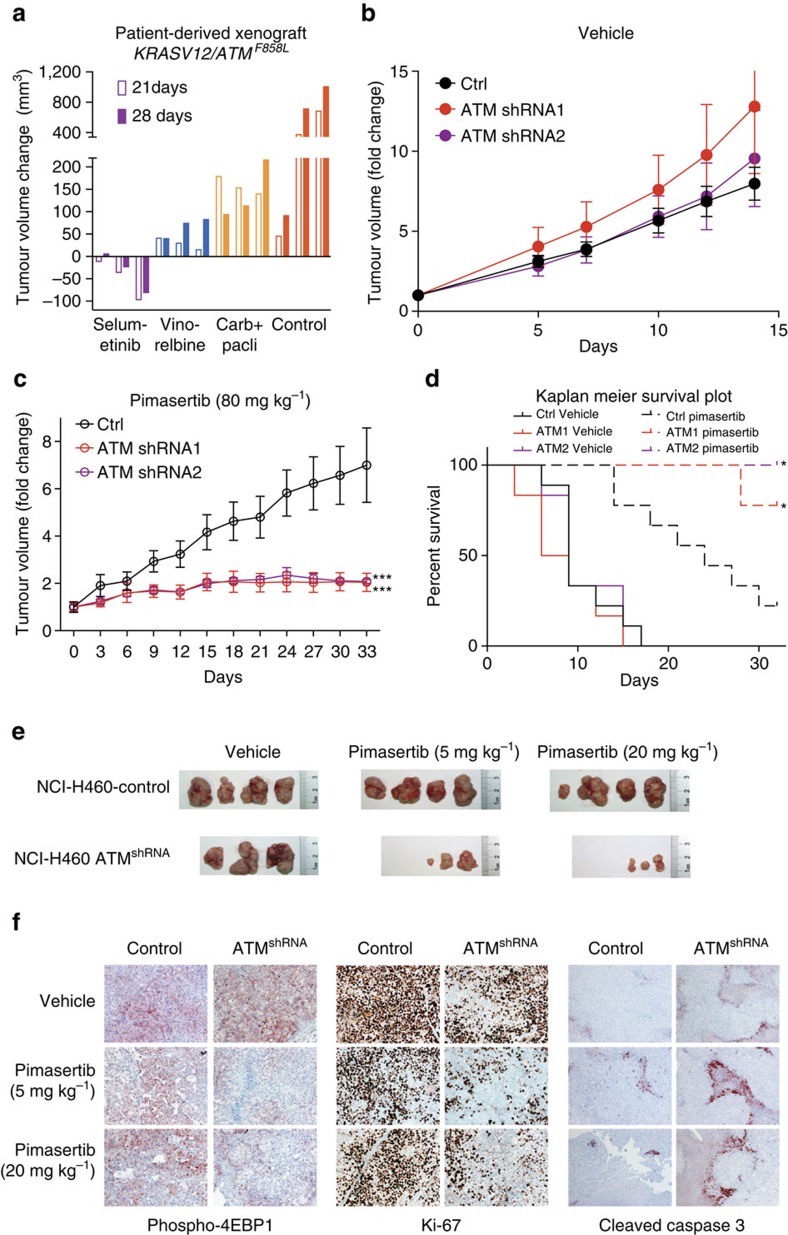
Lung cancer is the leading cause of cancer deaths, and effective treatments are urgently needed. Loss-of-function mutations in the DNA damage response kinase ATM are common in lung adenocarcinoma but directly targeting these with drugs remains challenging. Here we report that ATM loss-of-function is synthetic lethal with drugs inhibiting the central growth factor kinases MEK1/2, including the FDA-approved drug trametinib. Lung cancer cells resistant to MEK inhibition become highly sensitive upon loss of ATM both in vitro and in vivo. Mechanistically, ATM mediates crosstalk between the prosurvival MEK/ERK and AKT/mTOR pathways. ATM loss also enhances the sensitivity of KRAS- or BRAF-mutant lung cancer cells to MEK inhibition. Thus, ATM mutational status in lung cancer is a mechanistic biomarker for MEK inhibitor response, which may improve patient stratification and extend the applicability of these drugs beyond RAS and BRAF mutant tumours.
Figures
References
-
- Marshall E. Cancer research and the $90 billion metaphor. Science 331, 1540–1541 (2011). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
