

Cell Death in Chondrocytes, Osteoblasts, and Osteocytes
- PMID: 27929439
- PMCID: PMC5187845
- DOI: 10.3390/ijms17122045
Cell Death in Chondrocytes, Osteoblasts, and Osteocytes
Abstract
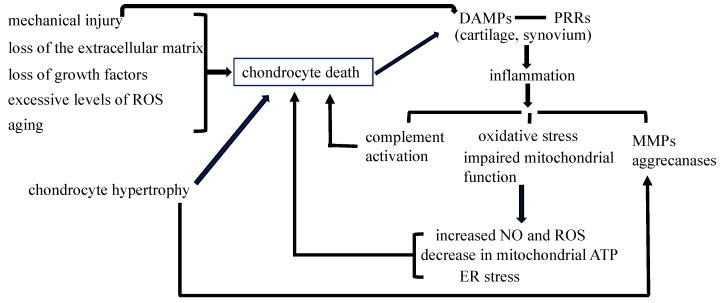
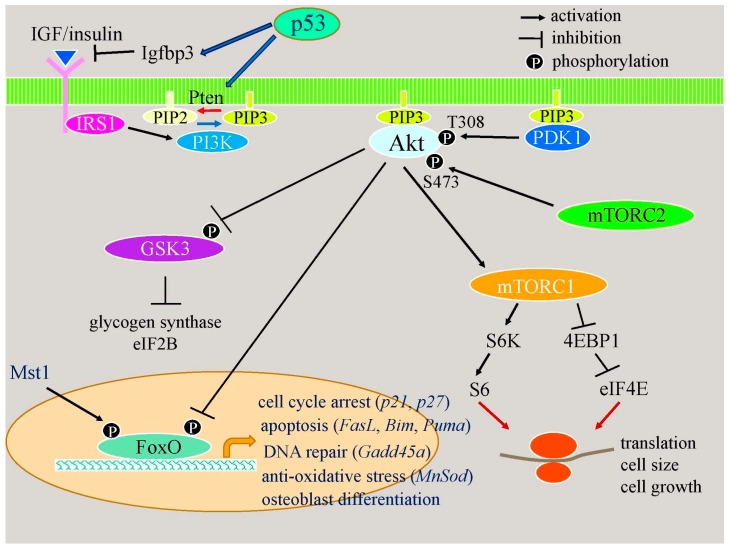
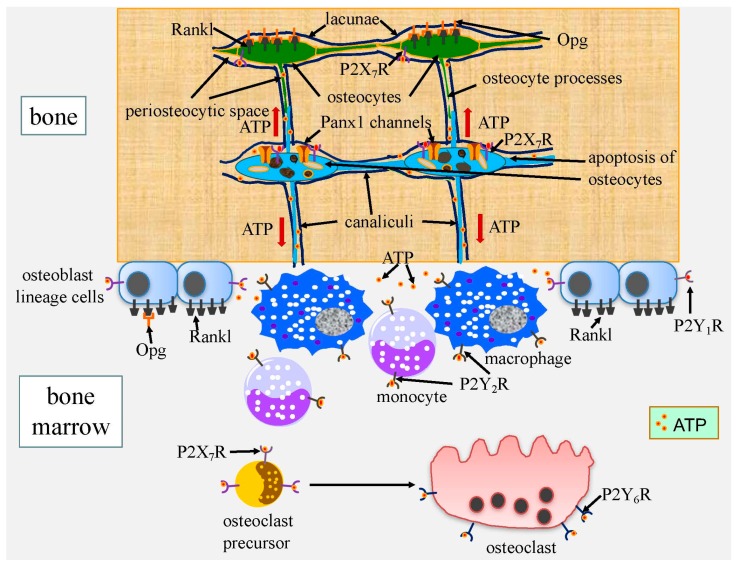
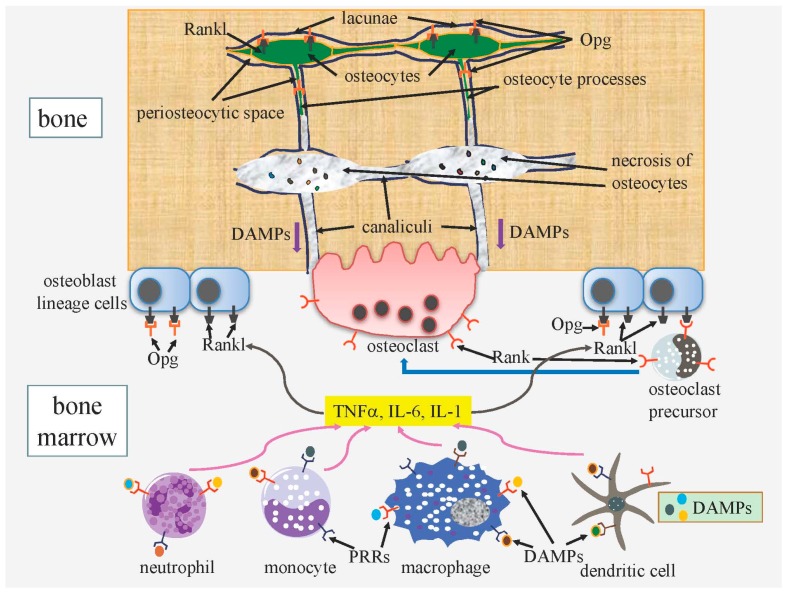
Cell death in skeletal component cells, including chondrocytes, osteoblasts, and osteocytes, plays roles in skeletal development, maintenance, and repair as well as in the pathogenesis of osteoarthritis and osteoporosis. Chondrocyte proliferation, differentiation, and apoptosis are important steps for endochondral ossification. Although the inactivation of P53 and RB is involved in the pathogenesis of osteosarcomas, the deletion of p53 and inactivation of Rb are insufficient to enhance chondrocyte proliferation, indicating the presence of multiple inhibitory mechanisms against sarcomagenesis in chondrocytes. The inflammatory processes induced by mechanical injury and chondrocyte death through the release of danger-associated molecular patterns (DAMPs) are involved in the pathogenesis of posttraumatic osteoarthritis. The overexpression of BCLXL increases bone volume with a normal structure and maintains bone during aging by inhibiting osteoblast apoptosis. p53 inhibits osteoblast proliferation and enhances osteoblast apoptosis, thereby reducing bone formation, but also exerts positive effects on osteoblast differentiation through the Akt-FoxOs pathway. Apoptotic osteocytes release ATP, which induces the receptor activator of nuclear factor κ-B ligand (Rankl) expression and osteoclastogenesis, from pannexin 1 channels. Osteocyte death ultimately results in necrosis; DAMPs are released to the bone surface and promote the production of proinflammatory cytokines, which induce Rankl expression, and osteoclastogenesis is further enhanced.
Keywords: ATP; BCLXL; DAMPs; FoxO; Rankl; Rb; apoptosis; necrosis; osteoarthritis; p53.
Conflict of interest statement
The author declares no conflict of interest.
Figures
References
-
- Yoshida C.A., Yamamoto H., Fujita T., Furuichi T., Ito K., Inoue K., Yamana K., Zanma A., Takada K., Ito Y., et al. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 2004;18:952–963. doi: 10.1101/gad.1174704. - DOI - PMC - PubMed
-
- Iwamoto M., Kitagaki J., Tamamura Y., Gentili C., Koyama E., Enomoto H., Komori T., Pacifici M., Enomoto-Iwamoto M. Runx2 expression and action in chondrocytes are regulated by retinoid signaling and parathyroid hormone-related peptide (PTHrP) Osteoarthr. Cartil. 2003;11:6–15. doi: 10.1053/joca.2002.0860. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
