

A piperazidine derivative of 23-hydroxy betulinic acid induces a mitochondria-derived ROS burst to trigger apoptotic cell death in hepatocellular carcinoma cells
- PMID: 27931237
- PMCID: PMC5146873
- DOI: 10.1186/s13046-016-0457-1
A piperazidine derivative of 23-hydroxy betulinic acid induces a mitochondria-derived ROS burst to trigger apoptotic cell death in hepatocellular carcinoma cells
Abstract
Background: Elevated production of reactive oxygen species (ROS) and an altered redox state have frequently been observed in hepatocellular carcinoma (HCC); therefore, selective killing of HCC cells by chemotherapeutic agents that stimulate ROS generation or impair antioxidant systems may be a feasible approach in HCC chemotherapy. Recently, betulinic acid and its derivatives have attracted attention because they showed anti-cancer effects via a ROS- and mitochondria-related mechanism. However, the source of ROS overproduction and the role of mitochondria were poorly identified, and the weak in vivo antitumour activity of these compounds limits their development as drugs.
Methods: Cytotoxicity was detected using MTT assays. In vivo anti-HCC effects were assessed using nude mice bearing HepG2 tumour xenografts. Cell cycle analysis, apoptosis rate and mitochondrial membrane potential were measured by flow cytometry. ROS production was detected using a microplate reader or a fluorescence microscope. Changes in gene and protein levels were measured by RT-PCR and western blotting, respectively. Other assays were performed using related detection kits.
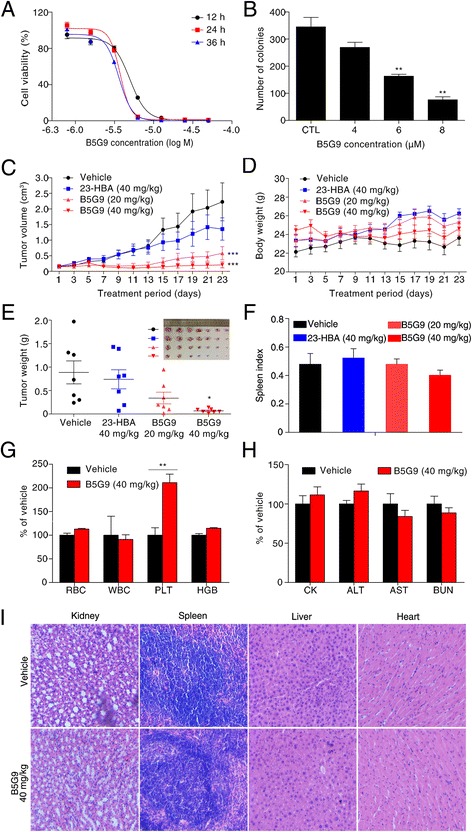
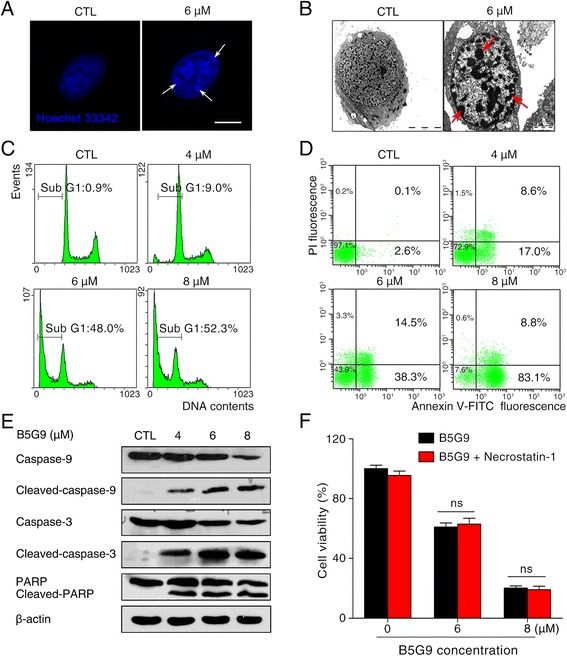
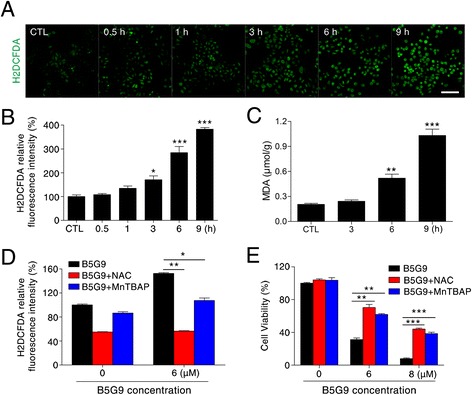
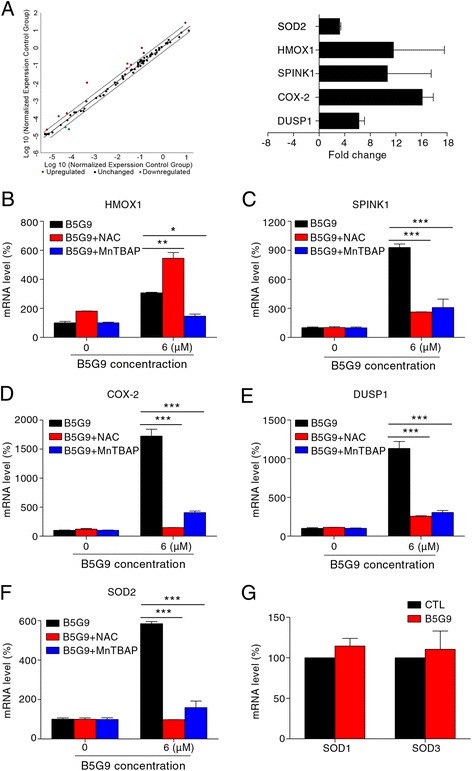
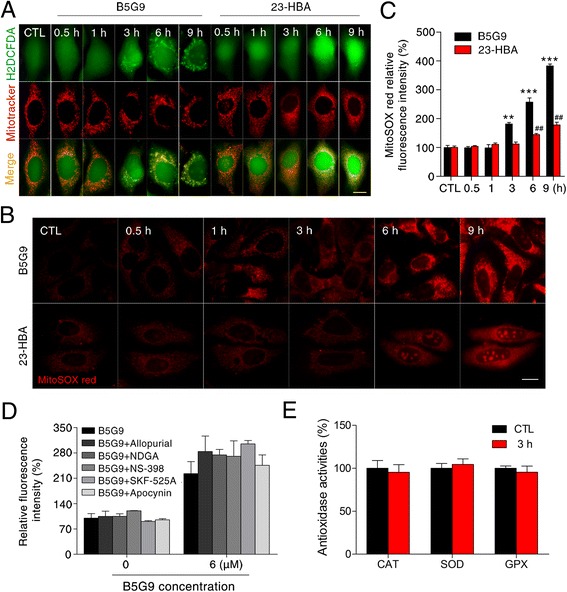
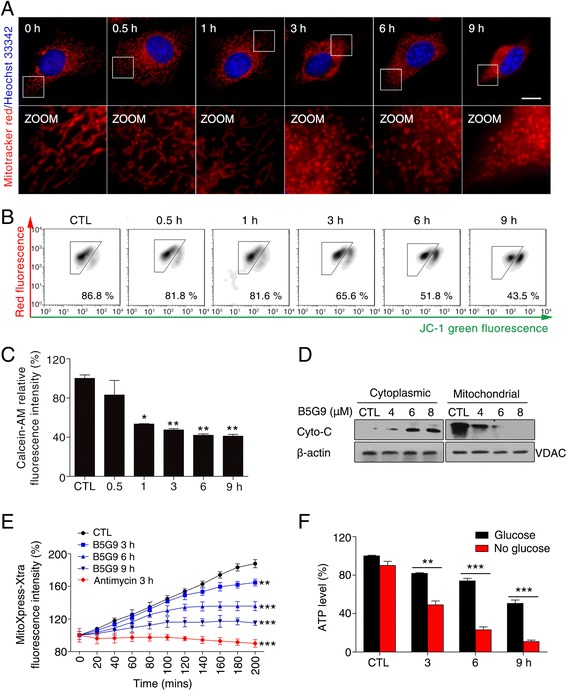
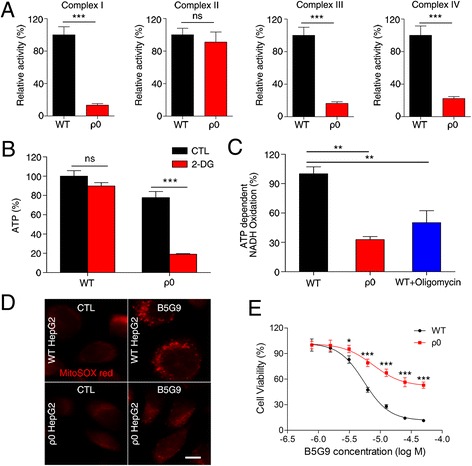
Results: B5G9, a piperazidine derivative of 23-hydroxy betulinic acid (23-HBA), showed excellent in vivo anti-HCC effects, with a tumour growth inhibitory rate of greater than 80%, and no significant side effects. B5G9 stimulated the production of ROS, which were derived from the mitochondria, but it had no effect on various other antioxidant systems. Moreover, B5G9 induced mitochondrial dysfunction, which was characterized by morphological changes, membrane potential collapse, membrane permeabilization, and decreases in the O2 consumption rate and ATP production. Furthermore, mtDNA-depleted ρ0 HepG2 cells were less sensitive to B5G9 treatment than wt HepG2 cells, indicating the importance of mitochondria in B5G9-induced cell death.
Conclusion: We discovered a piperazidine derivative of 23-HBA, B5G9, with excellent anti-HCC effects both in vivo and in vitro and no obvious toxic effects. The underlying mechanism was associated with mitochondria-derived ROS overproduction, and mitochondria played essential roles in B5G9-induced cell death. This study identified a potential agent for anti-HCC therapy and elucidated the mitochondria-related mechanism of BA and its derivatives.
Keywords: Apoptosis; Betulinic acid; Hepatocellular carcinoma; Mitochondria; Reactive oxygen species.
Figures
References
-
- Leung T, Johnson PJ. Systemic therapy for hepatocellular carcinoma. Cancer J. 2013;2:514–520. - PubMed
-
- Devasagayam TP, Tilak JC, Boloor KK, Sane KS, Ghaskadbi SS, Lele RD. Free radicals and antioxidants in human health: current status and future prospects. J Assoc Physicians India. 2004;52:794–804. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
