

The Long Noncoding RNA Transcriptome of Dictyostelium discoideum Development
- PMID: 27932387
- PMCID: PMC5295588
- DOI: 10.1534/g3.116.037150
The Long Noncoding RNA Transcriptome of Dictyostelium discoideum Development
Abstract
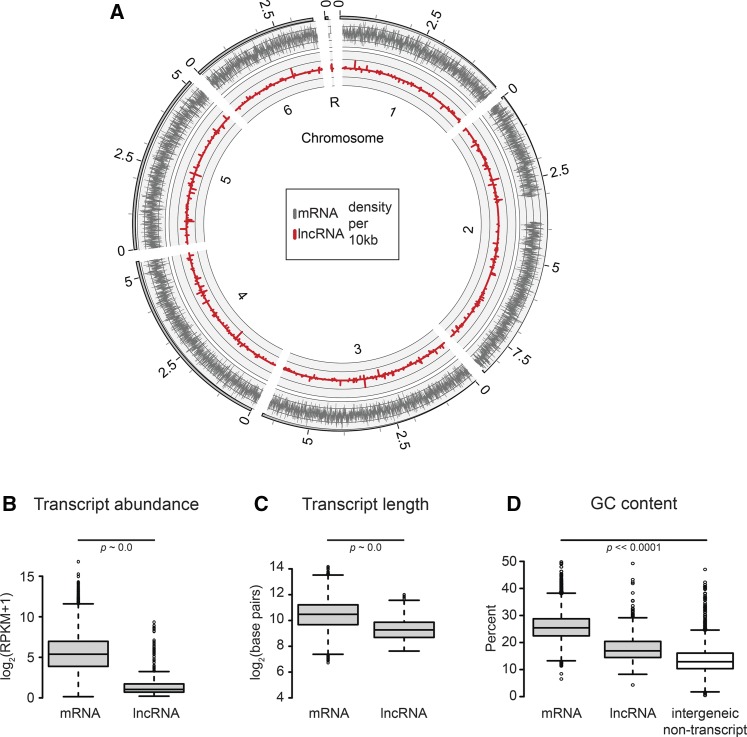
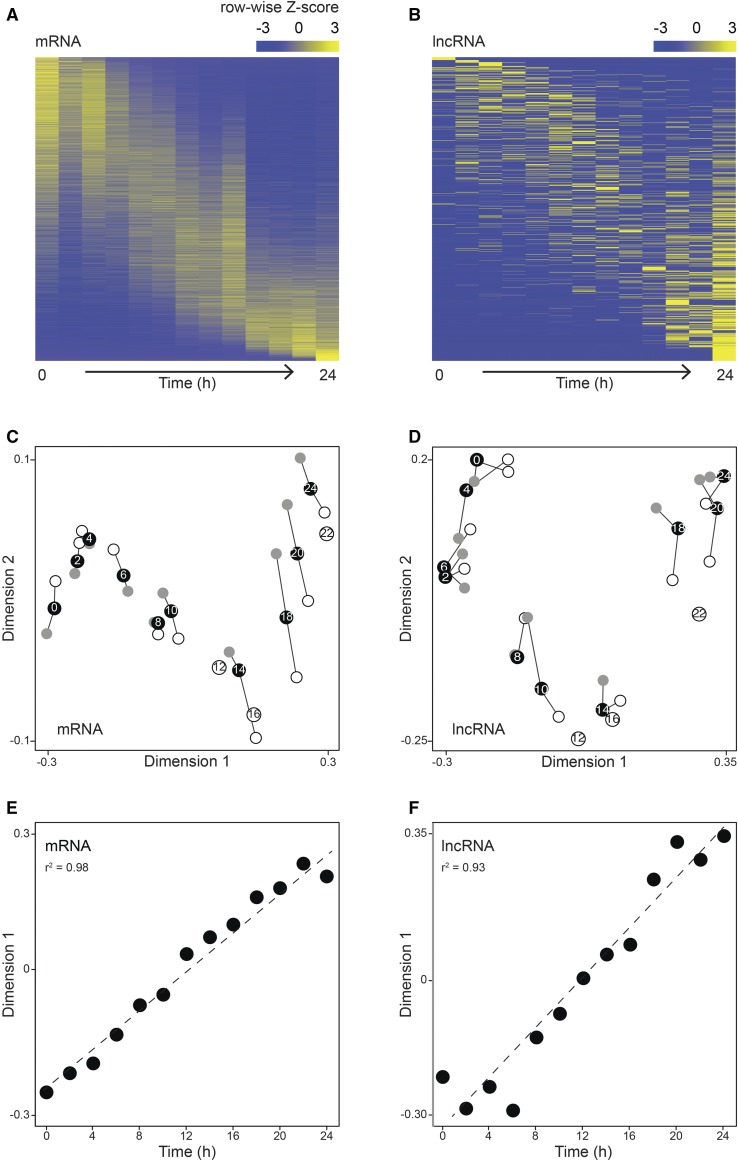
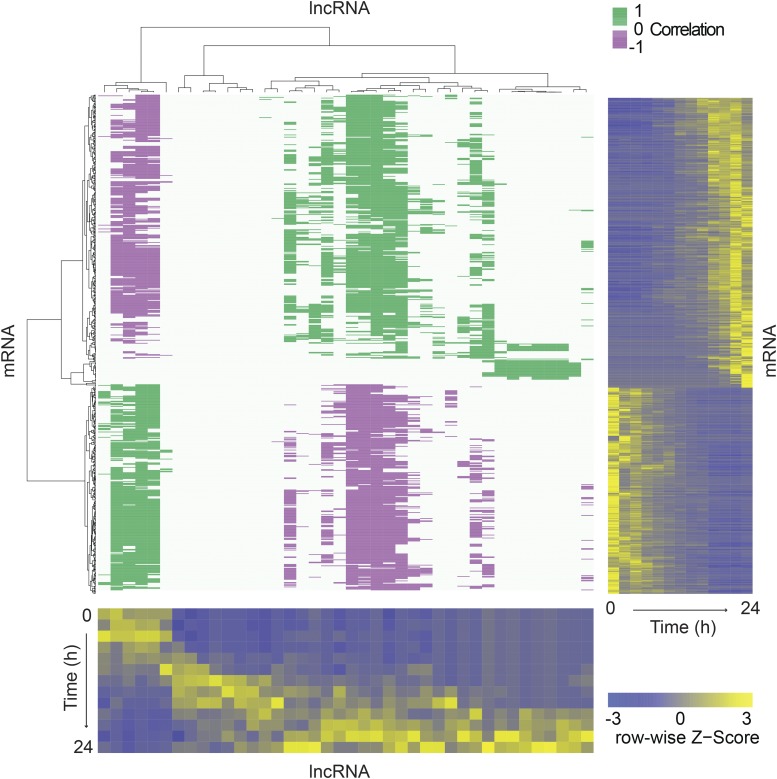
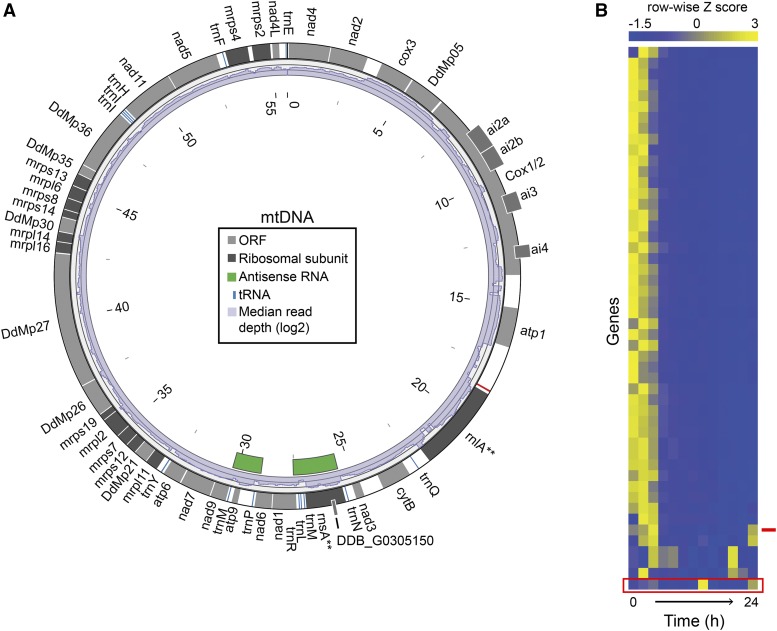
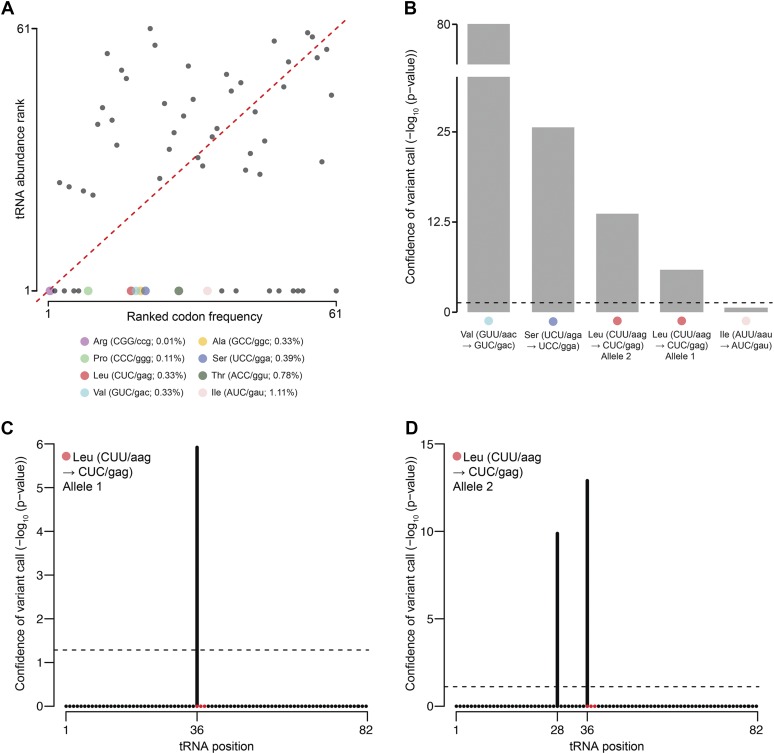
Dictyostelium discoideum live in the soil as single cells, engulfing bacteria and growing vegetatively. Upon starvation, tens of thousands of amoebae enter a developmental program that includes aggregation, multicellular differentiation, and sporulation. Major shifts across the protein-coding transcriptome accompany these developmental changes. However, no study has presented a global survey of long noncoding RNAs (ncRNAs) in D. discoideum To characterize the antisense and long intergenic noncoding RNA (lncRNA) transcriptome, we analyzed previously published developmental time course samples using an RNA-sequencing (RNA-seq) library preparation method that selectively depletes ribosomal RNAs (rRNAs). We detected the accumulation of transcripts for 9833 protein-coding messenger RNAs (mRNAs), 621 lncRNAs, and 162 putative antisense RNAs (asRNAs). The noncoding RNAs were interspersed throughout the genome, and were distinct in expression level, length, and nucleotide composition. The noncoding transcriptome displayed a temporal profile similar to the coding transcriptome, with stages of gradual change interspersed with larger leaps. The transcription profiles of some noncoding RNAs were strongly correlated with known differentially expressed coding RNAs, hinting at a functional role for these molecules during development. Examining the mitochondrial transcriptome, we modeled two novel antisense transcripts. We applied yet another ribosomal depletion method to a subset of the samples to better retain transfer RNA (tRNA) transcripts. We observed polymorphisms in tRNA anticodons that suggested a post-transcriptional means by which D. discoideum compensates for codons missing in the genomic complement of tRNAs. We concluded that the prevalence and characteristics of long ncRNAs indicate that these molecules are relevant to the progression of molecular and cellular phenotypes during development.
Keywords: Dictyostelium discoideum; development; noncoding RNA; ribosomal RNA depletion; slime mold; transcriptome time course.
Copyright © 2017 Rosengarten et al.
Figures
Similar articles
-
Leaps and lulls in the developmental transcriptome of Dictyostelium discoideum.BMC Genomics. 2015 Apr 13;16(1):294. doi: 10.1186/s12864-015-1491-7. BMC Genomics. 2015. PMID: 25887420 Free PMC article.
-
Long noncoding RNA repertoire in chicken liver and adipose tissue.Genet Sel Evol. 2017 Jan 10;49(1):6. doi: 10.1186/s12711-016-0275-0. Genet Sel Evol. 2017. PMID: 28073357 Free PMC article.
-
The transcriptome of Candida albicans mitochondria and the evolution of organellar transcription units in yeasts.BMC Genomics. 2015 Oct 21;16:827. doi: 10.1186/s12864-015-2078-z. BMC Genomics. 2015. PMID: 26487099 Free PMC article.
-
The Working Modules of Long Noncoding RNAs in Cancer Cells.Adv Exp Med Biol. 2016;927:49-67. doi: 10.1007/978-981-10-1498-7_2. Adv Exp Med Biol. 2016. PMID: 27376731 Review.
-
The specificity of long noncoding RNA expression.Biochim Biophys Acta. 2016 Jan;1859(1):16-22. doi: 10.1016/j.bbagrm.2015.08.005. Epub 2015 Aug 19. Biochim Biophys Acta. 2016. PMID: 26297315 Review.
Cited by
-
Lineage-Specific Genes and Family Expansions in Dictyostelid Genomes Display Expression Bias and Evolutionary Diversification during Development.Genes (Basel). 2021 Oct 16;12(10):1628. doi: 10.3390/genes12101628. Genes (Basel). 2021. PMID: 34681022 Free PMC article.
-
dictyExpress: a web-based platform for sequence data management and analytics in Dictyostelium and beyond.BMC Bioinformatics. 2017 Jun 2;18(1):291. doi: 10.1186/s12859-017-1706-9. BMC Bioinformatics. 2017. PMID: 28578698 Free PMC article.
-
Abundantly expressed class of noncoding RNAs conserved through the multicellular evolution of dictyostelid social amoebas.Genome Res. 2021 Mar;31(3):436-447. doi: 10.1101/gr.272856.120. Epub 2021 Jan 21. Genome Res. 2021. PMID: 33479022 Free PMC article.
-
Multiple developmental pathways in organisms with developmentally complex life cycles.Front Cell Dev Biol. 2025 May 14;13:1585073. doi: 10.3389/fcell.2025.1585073. eCollection 2025. Front Cell Dev Biol. 2025. PMID: 40438145 Free PMC article. Review.
-
Transcriptional milestones in Dictyostelium development.Genome Res. 2021 Aug;31(8):1498-1511. doi: 10.1101/gr.275496.121. Epub 2021 Jun 28. Genome Res. 2021. PMID: 34183452 Free PMC article.
References
-
- Andrews, S., 2010 FastQC: a Quality Control Tool for High Throughput Sequence Data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc. Accessed: June 7, 2016.
-
- Avesson L., Schumacher H. T., Fechter P., Romby P., Hellman U., et al. , 2011. Abundant class of non-coding RNA regulates development in the social amoeba Dictyostelium discoideum. RNA Biol. 8: 1094–1104. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases