

MAGI2 Mutations Cause Congenital Nephrotic Syndrome
- PMID: 27932480
- PMCID: PMC5407720
- DOI: 10.1681/ASN.2016040387
MAGI2 Mutations Cause Congenital Nephrotic Syndrome
Abstract
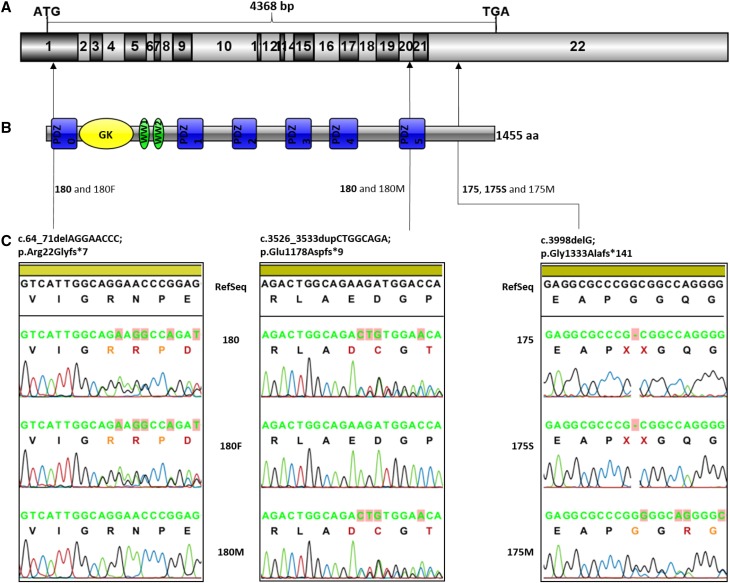
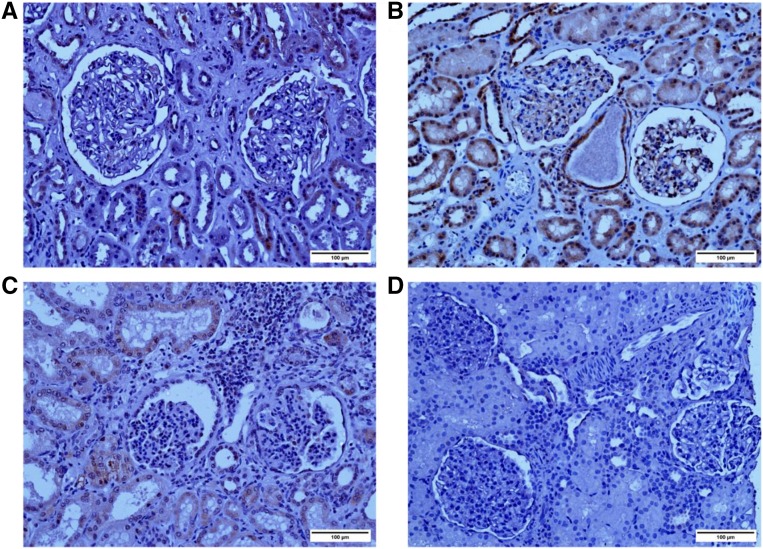
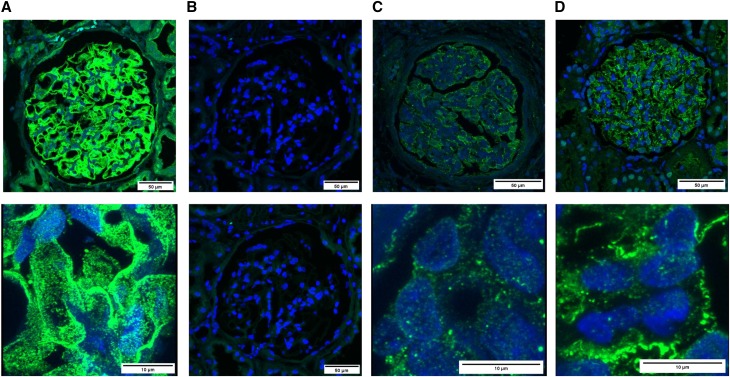
Steroid-resistant nephrotic syndrome (SRNS), a heterogeneous disorder of the renal glomerular filtration barrier, results in impairment of glomerular permselectivity. Inheritance of genetic SRNS may be autosomal dominant or recessive, with a subset of autosomal recessive SRNS presenting as congenital nephrotic syndrome (CNS). Mutations in 53 genes are associated with human SRNS, but these mutations explain ≤30% of patients with hereditary cases and only 20% of patients with sporadic cases. The proteins encoded by these genes are expressed in podocytes, and malfunction of these proteins leads to a universal end point of podocyte injury, glomerular filtration barrier disruption, and SRNS. Here, we identified novel disease-causing mutations in membrane-associated guanylate kinase, WW, and PDZ domain-containing 2 (MAGI2) through whole-exome sequencing of a deeply phenotyped cohort of patients with congenital, childhood-onset SRNS. Although MAGI2 has been shown to interact with nephrin and regulate podocyte cytoskeleton and slit diaphragm dynamics, MAGI2 mutations have not been described in human SRNS. We detected two unique frameshift mutations and one duplication in three patients (two families); two siblings shared the same homozygous frameshift mutation, whereas one individual with sporadic SRNS exhibited compound heterozygosity. Two mutations were predicted to introduce premature stop codons, and one was predicted to result in read through of the normal translational termination codon. Immunohistochemistry in kidney sections from these patients revealed that mutations resulted in lack of or diminished podocyte MAGI2 expression. Our data support the finding that mutations in the MAGI2 gene are causal for congenital SRNS.
Keywords: familial nephropathy; genetic renal disease; nephrin; nephrotic syndrome; podocyte; proteinuria.
Copyright © 2017 by the American Society of Nephrology.
Figures
References
-
- McCarthy HJ, Bierzynska A, Wherlock M, Ognjanovic M, Kerecuk L, Hegde S, Feather S, Gilbert RD, Krischock L, Jones C, Sinha MD, Webb NJ, Christian M, Williams MM, Marks S, Koziell A, Welsh GI, Saleem MA; RADAR the UK SRNS Study Group : Simultaneous sequencing of 24 genes associated with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 8: 637–648, 2013 - PMC - PubMed
-
- Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, Engelmann S, Vega-Warner V, Fang H, Halbritter J, Somers MJ, Tan W, Shril S, Fessi I, Lifton RP, Bockenhauer D, El-Desoky S, Kari JA, Zenker M, Kemper MJ, Mueller D, Fathy HM, Soliman NA, Hildebrandt F; SRNS Study Group : A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol 26: 1279–1289, 2015 - PMC - PubMed
-
- Miyake N, Tsukaguchi H, Koshimizu E, Shono A, Matsunaga S, Shiina M, Mimura Y, Imamura S, Hirose T, Okudela K, Nozu K, Akioka Y, Hattori M, Yoshikawa N, Kitamura A, Cheong HI, Kagami S, Yamashita M, Fujita A, Miyatake S, Tsurusaki Y, Nakashima M, Saitsu H, Ohashi K, Imamoto N, Ryo A, Ogata K, Iijima K, Matsumoto N: Biallelic mutations in nuclear pore complex subunit NUP107 cause early-childhood-onset steroid-resistant nephrotic syndrome. Am J Hum Genet 97: 555–566, 2015 - PMC - PubMed
-
- Braun DA, Sadowski CE, Kohl S, Lovric S, Astrinidis SA, Pabst WL, Gee HY, Ashraf S, Lawson JA, Shril S, Airik M, Tan W, Schapiro D, Rao J, Choi WI, Hermle T, Kemper MJ, Pohl M, Ozaltin F, Konrad M, Bogdanovic R, Büscher R, Helmchen U, Serdaroglu E, Lifton RP, Antonin W, Hildebrandt F: Mutations in nuclear pore genes NUP93, NUP205 and XPO5 cause steroid-resistant nephrotic syndrome. Nat Genet 48: 457–465, 2016 - PMC - PubMed
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
